基于粘性末端‑介导的链置换反应结合聚合切刻等温扩增技术检测UDG活性的方法与流程
本发明涉及一种基于粘性末端-介导的链置换反应结合聚合切刻等温扩增技术检测UDG活性的方法。
背景技术:
尿嘧啶-DNA糖基化酶(UDG)能对DNA中的尿嘧啶(U)损伤进行特异地识别和移除,进而引发U损伤的碱基移除修复过程(Stivers,J.T.;Jiang,Y.L.,A mechanistic perspective on the chemistry of DNA repair glycosylases.Chem.Rev.,2003,103,2729-2759;Parikh,S.S.;Walcher,G.;Jones,G.D.;Slupphaug,G.;Krokan,H.E.;Blackburn,G.M.;Tainer,J.A.,Uracil-DNA glycosylase-DNA substrate and product structures:conformational strain promotes catalytic efficiency by coupled stereoelectronic effects.PNAS.,2000,97,5083-5088.)。人体中,每天每个细胞的基因组中会出现数百个U损伤,它们一部分来自于胞嘧啶(C)的自发水解脱氨作用,另一部分来自于DNA复制过程中三磷酸脱氧尿苷酸(dUTP)的错误掺入(Sousa,M.M.;Krokan,H.E.;Slupphaug,G.,DNA-uracil and human pathology.Mol.Aspects Med.,2007,28,276-306;Kavli,B.;Sundheim,O.;Akbari,M.;Otterlei,M.;Nilsen,H.;Skorpen,F.;Aas,P.A.;Hagen,L.;Krokan,H.E.;Slupphaug,G.,hUNG2 is the major repair enzyme for removal of uracil from U:A matches,U:G mismatches,and U in single-stranded DNA,with hSMUG1 as a broad specificity backup.J.Biol.Chem.,2002,277,39926-39936.)。异常活性的UDG将不能对这些U损伤进行修复,从而引起基因突变,干扰基因组的完整性,导致相关疾病的发生(Bulgar,A.D.;Weeks,L.D.;Miao,Y.;Yang,S.;Xu,Y.;Guo,C.;Markowitz,S.;Oleinick,N.;Gerson,S.L.;Liu,L.,Cell Death Dis.,2012,3,e252.)。因此,灵敏检测UDG的活性能为功能研究和临床诊断提供一种有价值的策略。
目前检测UDG活性的常用方法需要放射性的标记或耗时的电泳过程(Ischenko,A.A.;Saparbaev,M.K.,Alternative nucleotide incision repair pathway for oxidative DNA damage.Nature,2002,415,183-187.)。为了实现安全且快速地检测UDG的活性,一些新型的生物传感方法已被发展,包括电化学生物传感法(McWilliams,M.A.;Anka,F.H.;Balkus,K.J.;Slinker,J.D.,Sensitive and selective real-time electrochemical monitoring of DNA repair.Biosens,Bioelectron.,2014,54,541-546.)、比色生物传感法(Liu,X.J.;Chen,M.Q.;Hou,T.;Wang,X.Z.;Liu,S.F.;Li,F.,Label-free colorimetric assay for base excision repair enzyme activity based on nicking enzyme assisted signal amplification.Biosens,Bioelectron.,2014,54,598-602.;Nie,H.;Wang,W.;Li,W.;Nie,Z.;Yao,S.,A colorimetric and smartphone readable method for uracil-DNA glycosylase detection based on the target-triggered formation of G-quadruplex.Analyst,2015,140,2771-2777.)以及荧光生物传感法(Liu,B.;Yang,X.;Wang,K.;Tan,W.;Li,H.;Tang,H.,Real-time monitoring of uracil removal by uracil-DNA glycosylase using fluorescent resonance energy transfer probes.Anal.Biochem.,2007,366,237-243.)。其中,荧光生物传感法已被广泛用于检测UDG的活性。为了进一步提高检测的灵敏度,信号扩增技术已被用于荧光生物传感方法的构建中。Yu课题组(Zhang,L.L.;Zhao,J.J.;Jiang,J.H.;Yu,R.Q.,A target-activated autocatalytic DNAzyme amplification strategy for the assay of base excision repair enzyme activity.Chem.Commun.,2012,48,8820-8822.)设计了一种含有多个U碱基的双链DNA探针,当UDG将该双链DNA探针中的多个U碱基移除之后,双链DNA发生解离并释放出DNA酶序列,从而引发DNA酶的自催化信号扩增过程。
申请号为CN201510061929.X的专利公开了一种基于DNA机器的荧光信号扩增方法用于UDG的活性检测,其中DNA机器的引发部分也是一种含有多个U碱基的双链DNA探针,并且只有当该双链DNA探针中的多个U碱基都被UDG移除之后,它才能释放出引物序列来引发DNA机器的运行。在这些信号扩增型的荧光生物传感方法中,它们通过直接杂交模式将信号探针或引物序列封闭在双链DNA中,并把这种含多个U碱基的双链DNA作为UDG的识别探针。然而,当低活性的UDG只能移除探针中的1个或2个U碱基而产生少量无嘌呤/无嘧啶位点(AP)位点时,由于AP位点也是一种错配类型,而直接杂交模式难以辨别少量错配,因此信号探针或引物序列此时仍被封闭在双链DNA中,导致响应减弱,限制了灵敏度的提高。
技术实现要素:
本发明的目的之一是提供一种检测UDG活性的探针,目的之二是提供一种基于粘性末端-介导的链置换反应结合聚合切刻等温扩增技术检测UDG活性的方法,目的之三是提供一种检测UDG活性的试剂盒。
具体技术方案如下:
一种检测UDG活性的探针,包括单链DNA识别探针UP、发夹探针TP以及信号报道探针RP;
所述单链DNA识别探针包括:引物序列、1个或两个连续的U碱基,所述两个连续的U碱基在单链DNA的端部;
所述发夹探针TP包括,粘性末端、与识别探针UP完全互补的颈部碱基序列;
所述信号报道探针RP,是一种发夹结构,3’端含有悬垂的序列,所述悬垂序列能够与识别探针UP序列杂交,颈部含有G4DNA的互补序列和切刻酶Nt.BstNBI的识别序列。
一种检测UDG活性的方法,基于粘性末端-介导的链置换反应结合聚合切刻等温扩增技术,步骤如下:
(1)将发夹探针TP以及信号报道探针RP加热变性降温后备用;
(2)对UDG的识别反应:将识别探针UP与待测的UDG加入到1×ThermoPol缓冲溶液中进行孵育反应,;
(3)TSDR对产物AP错配的特异性识别:向步骤(2)的反应产物中加入发夹探针TP和10×ThermoPol缓冲溶液进行孵育反应;
(4)聚合切刻等温扩增反应:向步骤(3)反应产物中加入1×ThermoPol缓冲溶液,NEB buffer 3,dNTPs,信号报道探针RP,Vent(exo-)DNA聚合酶以及Nt.BstNBI切刻酶混合反应;
(5)向步骤(4)反应产物中加入KCl溶液,NMM溶液,混合反应,对反应产物进行荧光测定,构建荧光强度与UDG浓度之间的线性曲线,对某一样品通过测定样品的荧光强度,代入线性曲线,计算UDG浓度;
所述识别探针UP、发夹探针TP、信号报道探针RP同上所述。
一种基于粘性末端-介导的链置换反应结合聚合切刻等温扩增技术检测UDG活性的方法,步骤如下:
(1)将含有粘性末端的发夹探针TP以及信号报道探针RP加热变性(优选:置于90℃的条件下加热变性5min),待温度缓慢降至室温,将所得产物保存备用(优选:置于4℃的条件下保存备用);
(2)将100nM-500nM识别探针UP与待测的UDG加入到1×ThermoPol缓冲溶液(20mM Tris-HCl,pH 8.8,10mM KCl,10mM(NH4)2SO4,2mM MgSO4,0.1%TritonX-100)中,得到混合溶液,并将该溶液混匀后孵育反应(优选:37℃的条件下孵育20min-60min),以完成对UDG的识别反应;
(3)向步骤(2)的反应产物中加入150nM-1.80μM发夹探针TP和10×ThermoPol缓冲溶液(200mM Tris-HCl,pH 8.8,100mM KCl,100mM(NH4)2SO4,20mM MgSO4,1%TritonX-100),得到混合溶液,并将该溶液下孵育反应(优选:37℃的条件下反应30min-120min),以完成TSDR对产物AP错配的特异性识别;
(4)向步骤(3)反应产物中加入1×ThermoPol缓冲溶液,NEB buffer 3,0.05mM-0.50mM的dNTPs,150nM-375nM的信号报道探针RP,0.02U/μL-0.20U/μL的Vent(exo-)DNA聚合酶以及0.05U/μL-0.50U/μL的Nt.BstNBI切刻酶,得到混合溶液,混匀后反应(优选:混匀后将其置于55℃的条件下反应30min-100min),以进行聚合切刻等温扩增反应;
(5)向步骤(4)反应产物中加入160mM的KCl,1.00μM-10.0μM的NMM至终体积为60μL,并将该混合物反应(优选:37℃条件下孵育10min-60min),对反应产物进行荧光测定,构建荧光强度与UDG浓度之间的线性曲线,对某一样品通过测定样品的荧光强度,代入线性曲线,计算UDG浓度。
一种检测UDG活性的试剂盒,包括上述的单链DNA识别探针UP、发夹探针TP以及信号报道探针RP。所述的试剂盒,还包括1×ThermoPol缓冲溶液、10×ThermoPol缓冲溶液、NEB buffer 3、Vent(exo-)DNA聚合酶、Nt.BstNBI切刻酶。
上述方法和探针在检测HELA细胞裂解液中的UDG活性中的应用。
基于粘性末端-介导的链置换反应结合聚合切刻等温扩增方法对UDG活性进行检测的原理:
以TSDR来对少量U碱基的移除进行响应,并结合聚合切刻等温扩增技术,发展了一种荧光信号扩增方法用于灵敏检测UDG的活性,具体的设计原理如图1所示。识别探针UP是一条单链DNA,它含有引物序列和两个U碱基。发夹探针TP的3’端含有粘性末端序列,并且该粘性末端以及与之相连的颈部的序列能与识别探针UP完全互补。另外,信号报道探针RP也是一种发夹结构,它的3’端含有悬垂的序列,颈部含有G4 DNA的互补序列,并且环部还含有切刻酶Nt.BstNBI的识别序列。在UDG的作用下,识别探针UP中的两个U碱基被移出,从而生成一条含有两个AP位点的UP’探针。由于AP错配能抑制UP’与TP中粘性末端的杂交,因此TSDR将不会发生,从而使得单链探针UP’中的引物序列仍能自由存在。随后,该引物序列能与信号报道探针RP中的悬垂序列发生杂交,并在聚合酶Vent(exo-)的作用下引发链延伸反应,得到DNA双链结构。接着,Nt.BstNBI识别并切刻双链DNA中完整的识别位点,产生的切口又能作为聚合酶作用的新位点,进而继续引发链延伸反应。通过循环的聚合-切刻-置换反应,系统释放出大量的G4序列,它们在单价阳离子的存在下能折叠成G4结构,并与NMM发生特异性地结合,产生增强的荧光信号。然而,当UDG不存在时,单链探针UP能与发夹探针TP中的粘性末端发生杂交,进而引发TSDR,导致探针UP中的引物序列被封闭,从而不能引发后续的等温扩增反应。本方法能通过TSDR来对少量U碱基的移除进行响应,并结合聚合切刻等温扩增技术,以实现对UDG活性的灵敏检测。
本发明的有益效果:
本发明以TSDR来对少量U碱基的移除进行响应,并结合聚合切刻等温扩增技术,构建了一种荧光信号扩增方法用于灵敏检测UDG的活性。在本方法中,分别设计了单链DNA识别探针、含有粘性末端的发夹探针以及信号报道探针。当UDG存在时,单链DNA探针中的两个U碱基被移除,得到含有AP错配的单链DNA探针。基于TSDR对少量错配的特异性识别能力,上述含AP错配的单链DNA探针将不能与含粘性末端的发夹探针发生TSDR,从而使单链DNA探针中的引物序列仍能自由存在。随后,该引物序列与信号报道探针发生杂交,进而引发后续的聚合切刻等温扩增反应,实现了对少量U碱基移除的灵敏响应,提高了检测UDG活性的灵敏度。然而,当UDG不存在时,单链DNA探针能与含粘性末端的发夹探针发生TSDR,导致引物序列被封闭,从而不能引发后续的等温扩增反应,降低了阴性信号。本方法能检测低至0.000027U/mL的UDG活性。
附图说明
图1为本发明方法检测UDG活性的原理图;
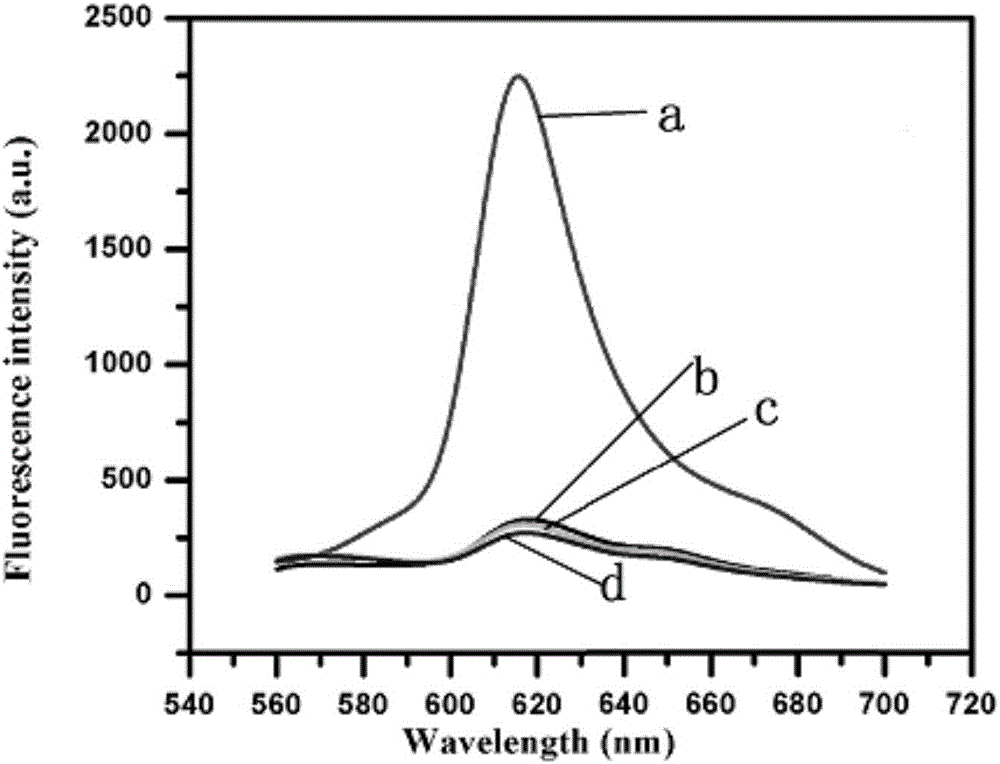
图2为在不同条件下,传感系统的荧光发射光谱:a:UP+UDG+TP+RP,b:UP+TP+RP,c:UDG+TP+RP,d:UP+UDG+TP;
图3ΔF相对于UDG浓度的标准曲线,内插图为传感系统对低浓度UDG活性的线性响应,误差棒为三次平行实验结果的标准偏差;
图4在不同条件下,考察传感系统的特异性:(1)不含UDG的对照实验,(2)UDG,(3)混合样品(UDG,hOGG1,hAAG和DNase I),(4)hOGG1,(5)hAAG,(6)DNase I,各酶的浓度均为1.0U/mL。误差棒为三次平行实验结果的标准偏差;
图5传感系统对HeLa细胞裂解液的荧光光谱响应以及UGI对HeLa细胞裂解液中UDG活性的抑制效果。
具体实施方式
下面结合附图与实施例对本发明作进一步说明。
本发明实施例中所用到的实验试剂和仪器如下:
本发明使用的DNA寡核苷酸是由生工(中国,上海)合成和纯化的,(TP探针:TGGCGGAGGCAGGAGTTTTTTTTTTTCTCCTGCCTCCGCCAAATTGT;UP探针:ACAAUUTGGCGGAGGCAGGAG;RP探针:CCCAACCCGCCCTACCCTTTTTGACTCGTAGGGCGGCTCCTGCCTCCGCCA),其中斜体代表悬垂序列,标灰字母代表尿嘧啶脱氧核糖核苷酸。UDG,8-氧化鸟嘌呤DNA糖化酶(hOGG1),烷基化腺嘌呤DNA糖基化酶(hAAG),DNA酶I(DNase I),尿嘧啶糖基化酶抑制剂(UGI),Vent(exo-)DNA聚合酶,Nt.BstNBI切刻酶都是购于New England Biolabs(中国,北京)。一单位的UDG是指在1min内催化水解双链DNA中60pmol的U碱基所需要的酶量。一单位的UGI是指在体积为50μL的反应体系中1h内抑制一个单位的UDG所需要的蛋白量。三磷酸脱氧核苷酸(dNTPs)购于Fermentas(中国,北京)。NMM购于Frontier Scientific(美国,犹他州,罗根)。二甲基亚砜(DMSO)是从生工(中国,上海)购得的。所有其它的化学药品都是分析纯的。制备溶液时用到的超纯水都是来自Millipore Milli-Q水净化系统(>18.25MΩ·cm)。
所有的荧光测试都是在日立F-7000荧光分光光度计(日本,日立公司)上进行的。激发波长为399nm,收集的发射波长的范围是560nm-700nm。我们利用618nm处的荧光强度来评价本方法的分析性能。激发狭缝宽度和发射狭缝宽度都被设置为10nm,光电倍增管电压被设置为700V。
实施例1检测UDG的活性
为了得到相应的发夹结构,分别将含有粘性末端的发夹探针TP以及信号报道探针RP置于90℃的条件下加热变性5min,待温度缓慢降至室温,将所得产物置于4℃的条件下保存备用。将100nM-500nM识别探针UP与不同浓度的UDG加入到1×ThermoPol缓冲溶液(20mM Tris-HCl,pH 8.8,10mM KCl,10mM(NH4)2SO4,2mM MgSO4,0.1%TritonX-100)中,得到终体积为20μL的混合溶液,并将该溶液混匀后置于37℃的条件下孵育20min-60min,以完成对UDG的识别反应。接着,向上述反应产物中加入150nM-1.80μM的发夹探针TP和1.0μL 10×ThermoPol缓冲溶液(200mM Tris-HCl,pH 8.8,100mM KCl,100mM(NH4)2SO4,20mM MgSO4,1%TritonX-100),得到终体积为30μL的混合溶液,并将该溶液置于37℃的条件下反应30min-120min,以完成TSDR对产物AP错配的特异性识别。随后,向上述反应产物中加入1×ThermoPol缓冲溶液,NEB buffer 3,0.05mM-0.50mM的dNTPs,150nM-375nM的信号报道探针RP,0.02U/μL-0.20U/μL的Vent(exo-)DNA聚合酶以及0.05U/μL-0.50U/μL的Nt.BstNBI切刻酶,得到混合溶液的终体积为50μL,混匀后将其置于55℃的条件下反应30min-100min,以进行聚合切刻等温扩增反应。最后,向上述反应产物中加入160mM的KCl,1.00μM-10.0μM的NMM至终体积为60μL,并将该混合物置于37℃条件下孵育10min-60min。
实施例2本发明方法检测Hela细胞裂解液中的UDG活性
制备Hela细胞裂解液:Hela细胞样品通过离心分离(5min,3000rpm,4℃)而被压成丸状,并在冰上通过超声波仪而重新分散在裂解缓冲液(10mM Tris-Hcl,pH 7.0)中。混合溶液随后在4℃下以12,000rpm的速度进行离心分离30min以移除不可溶的物质。收集上清液并通过0.45μm的滤膜过滤,产生粗制的Hela细胞裂解液。
将1.0μL的HeLa细胞裂解液作为待测样品,其中该HeLa细胞裂解液的浓度为每1.0μL细胞裂解液中含1.0×104个细胞。如图5所示,缓冲溶液仅能引起非常低的荧光信号,而HeLa细胞裂解液则能引起明显增强的荧光信号。另外,当向含有HeLa细胞裂解液的系统中加入UDG的抑制剂UGI之后,传感系统的荧光强度仅与缓冲溶液的相当。这表明,传感系统荧光信号的增强是由HeLa细胞裂解液中的UDG引起的。并且,本方法对细胞裂解液中其它组分的干扰具有耐受性。
实施例3对UDG活性进行检测的可行性验证
利用荧光发射光谱验证了本方法用于检测UDG活性的可行性。如图2中的曲线d所示,当传感系统中不含信号报道探针RP时,传感系统表现出非常低的荧光信号。这表明荧光信号的产生离不开探针RP的存在。如图2中的曲线c所示,当系统中不含识别探针UP时,传感系统的荧光信号也非常低。这表明在本方法中,传感系统对UDG的识别离不开UP探针的参与。如图2中的曲线b所示,当UDG不存在时,传感系统仍表现出非常低的荧光信号。这是由于探针UP能够与发夹探针TP发生TSDR,使探针UP中的引物序列被封闭,导致后续的信号扩增及产生过程不能进行。如图2中的曲线a所示,当UDG存在时,传感系统则表现出明显增强的荧光信号。这表明当UDG存在时,探针UP中的U碱基被移除而形成含有AP错配的UP’探针,其中AP错配将抑制UP’与TP探针之间发生TSDR,这使得UP’探针中的引物序列仍能自由存在,该引物序列能引发随后的信号扩增及产生过程。上述结果验证了本方法用于检测UDG活性的可行性。
实施例4检测UDG的活性
为了定量检测UDG的活性,在最优反应条件下对不同浓度的UDG活性进行了检测。图3展示了传感系统的净信号强度ΔF与UDG浓度的标准曲线,并且内插图显示出本方法对UDG活性的线性检测范围是从0.00020U/mL到0.0080U/mL,线性回归方程为ΔF=30.13+9.936×104CUDG,R2=0.9993。根据3δ原则,本方法对UDG活性的检测限为0.000027U/mL的,这低于目前文献报道的检测限。
实施例5本发明方法在检测UDG的活性时的精密度和重现性
另外,研究了本方法检测UDG活性的精密度和重现性。为了考察本方法的精密度,在同一天内,对目标样品进行了一系列的三次平行测定。对于UDG浓度分别为0.001U/mL,0.004U/mL和0.006U/mL的样品,得到的相对标准偏差(RSD)分别为4.1%,3.7%和3.1%。另外,为了考察本方法的重现性,在三个不同的日子里,对目标样品进行了一系列的三次平行测定。对于含不同浓度(0.001U/mL,0.004U/mL和0.006U/mL)UDG的样品,RSD值分别为5.8%,5.9%和5.5%。这些结果表明本方法在检测UDG的活性时具有合格的精密度和重现性。
实施例6本发明本方法在检测UDG的活性时的特异性
为了考察本方法的特异性,研究了传感系统对UDG以及其他酶(包括hOGG1,hAAG和DNase I)的荧光响应,结果见图4。只有UDG能引起明显增强的荧光信号,而hOGG1,hAAG和DNase I均不能引起荧光信号的增强。另外,当UDG与hOGG1,hAAG和DNase I混合之后,混合物中的UDG仍能引起荧光信号的增强,并且此时的信号强度与UDG单独存在时引起的信号强度相当。这表明本方法能特异性地检测UDG的活性。
上述虽然结合附图对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 还没有人留言评论。精彩留言会获得点赞!