一种基于CRISPR-Cas9技术的miR-126全长基因敲除试剂盒及其应用的制作方法
本发明属于基因工程领域,涉及一种miR-126基因敲除试剂盒,特别涉及一种基于CRISPR-Cas9技术可以对miR-126全长基因序列进行特异性完全敲除的试剂盒。此外,本发明还公开了该试剂盒的应用。
背景技术:
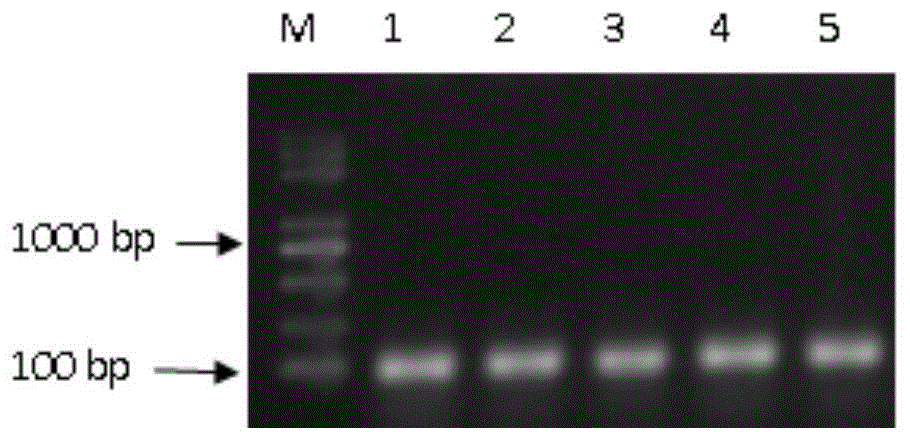
:microRNA(微RNA)是一类由内源基因编码的长度约为22个核苷酸的非编码单链RNA分子,广泛分布在病毒、植物到高等哺乳动物中,其在细胞内具有多种重要的调节作用。每个microRNA可以有多个靶基因,而几个microRNA也可以调节同一个基因。这种复杂的调节网络既可以通过一个microRNA来调控多个基因的表达,也可以通过几个microRNA的组合来精细调控某个基因的表达,因此microRNA的基因功能探索和应用开发一直是研究热点。miR-126(microRNA126,微RNA126)位于人类染色体9q34.3的类表皮生长因子域7(epidermalgrowthfactorlike7,EFGL7)基因的内含子中。miR-126是一种血管内皮细胞特异性表达的microRNA,高度表达于血管形成丰富的组织,如心脏、肺脏、肾脏中。它能够通过调控Spred-1(Sprouty-related,EVH1domain-containingprotein1,Sprouty相关含EVH1区域蛋白1)、VCAM-1(Vascularcelladhesionprotein1,血管细胞粘附分子-1)、HoxA9(homeoboxA9,同源盒基因A9)、v-Crk(SarcomaVirusCT10regulatorofkinase,肉瘤病毒CT10调节激酶)、EGFL-7和VEGF(vascularendothelialgrowthfactor,血管内皮生长因子)等基因的表达参与调节血管发育、新生血管形成以及血管炎症反应等血管的生理和病理生理过程。经其他研究发现,miR-126在胃肠道,生殖道癌症,乳腺、甲状腺、肺、以及其他一些癌症中表达被抑制。下调miR-126后可诱导癌细胞增殖,迁移,侵袭以及影响细胞存活期,miR-126下调与多种癌症生存不良之间存在相关性。这些研究发现为胃肠道,生殖道癌症,乳腺、甲状腺、肺等癌症转移预防判断提供了潜在的靶标,并为胃肠道,生殖道癌症,乳腺、甲状腺、肺等癌症的生物治疗指明新方向,提示miR-126研究的重要性和必要性。由于microRNA序列较短且其前体具有茎环结构,经典的RNA干扰(RNAi)的方法不适用于microRNA下调表达的功能研究。目前microRNA研究常用的方法,是应用生物学技术在体内细胞模型或体外动/植物模型中,将microRNA进行序列封闭或者造成基因缺失突变,模拟microRNA功能缺失引起的负向调节作用弱化或撤除,进而研究其功能作用机理,技术方法主要有两类:第一类方法是设计合成microRNA的互补封闭序列,互补序列与microRNA原来特定结合的mRNA形成竞争,弱化microRNA对特定mRNA结合能力和调控作用。由于互补序列是以碱基互补的形式对microRNA进行封闭,并不直接导致microRNA降解或表达的下调,所以microRNA的表达水平不发生变化,且没有直接的技术方法可以对microRNA功能的封闭效率进行量化检测,不能直接证明microRNA的功能受到了屏蔽,只能通过检测microRNA可能的调控下游基因变化来间接说明microRNA的功能受到抑制,因而此方法对microRNA功能研究的真实性和有效性存在较大的影响。第二类方法是用转录激活样效应因子核酸酶(transcriptionactivator-likeeffectornuclease,TALEN)TALEN技术,通过DNA识别模块将TALEN元件靶向特异性的DNA位点并结合,然后在FokI核酸酶的作用下完成特定位点的剪切,可以对microRNA进行DNA水平的剪切和修饰,模拟microRNA的功能缺失。但TALEN技术对目标基因的敲除依赖DNA结合的氨基酸聚合体模块序列,单个的TALEN模块进行组装需要大量的分子克隆和测序操作,十分繁琐,若所有工作在实验室内部完成基本实验操作需要耗时2个月以上,若筛选稳定敲除基因的细胞系需4个月以上,整个过程耗时长,成本高。CRISPR(ClusteredRegularlyInterspacedShortPalindromicRepeats/cas,成簇的、规律间隔的短回文重复序列)技术又称为CRISPR/Cas9(CRISPRassociatedprotein9,CRISPR相关蛋白9)核酸酶技术是最新出现的一种基因组编辑工具,它能够完成RNA导向的DNA识别及编辑。与上述介绍的基因技术工具相比,CRISPR技术更易于操作,具有更强的可扩展性。CRISPR/Cas9最早是在细菌的天然免疫系统内发现,其主要功能是对抗入侵的病毒及外源DNA,是细菌和古细菌的获得性免疫防御机制。在这一系统中,crRNA(CRISPR-derivedRNA,CRISPR衍生RNA)通过碱基配对与tracrRNA(trans-activatingRNA,反式激活RNA)结合形成双链RNA,此tracrRNA/crRNA二元复合体指导Cas9核酸酶蛋白在crRNA引导序列的靶定位点剪切双链DNA,从而达到对基因组DNA进行剪切或修饰的目的。为了实现在哺乳动物中应用CRISPR/Cas9系统,科学家对CRISPR/Cas9进行改造,将tracrRNA/crRNA二元复合体融合表达形成一条单链的导向RNA。而在实际设计中,只设计合成识别靶序列的一段20nt(碱基)左右的RNA序列并称之sgRNA(smallguideRNA,小向导RNA),其他部分导向RNA因为序列不变而作为sgRNA表达载体上的载体序列存在。因此,广泛应用的CRISPR/Cas9系统变为由含sgRNA的导向RNA指导下的Cas9核酸酶对靶向基因进行定向编辑的技术。CRISPR/Cas9技术的定点剪切编辑技术最常用的是针对单个位点设计sgRNA引导Cas9在靶标序列上进行定向剪切(例如,中国发明专利申请CN105112445A公开的“一种基于CRISPR-Cas9基因敲除技术的miR-205基因敲除试剂盒”),切割后细胞内固有的非同源末端连接途径(NHEJ)修复过程,会引入随机插入和删除的基因突变,这个修复过程不受人为控制,导致各类不必要的和不准确的序列插入和删除,经常产生不明确或无效的细胞表型,对研究产生不利影响。目前,尚未见有关利用CRISPR/Cas9系统特异性地将miR-126的全长基因序列进行敲除的报道。技术实现要素:本发明要解决的技术问题之一是提供一种基于CRISPR/Cas9基因编辑技术对miR-126基因进行基因敲除的试剂盒,其使得CRISPR/Cas9可以对靶标miR-126基因编码序列两端同时进行定点的剪切,能极大地避免采用传统的针对单个位点设计sgRNA引导Cas9在靶标序列上进行定向剪切所导致的非同源末端连接途径NHEJ引入随机突变发生的可能性,非常有利于产生针对于该基因完全删除的单一且明显的细胞表型,该试剂盒能高效、快速、精确、稳定地实现miR-126全长编码序列在基因组水平上完整敲除。本发明要解决的技术问题之二是提供该试剂盒的应用,该试剂盒可应用于多种细胞类型(例如,血管内皮细胞,以及多种恶性肿瘤如:乳腺癌、肺癌等)的miR-126基因敲除、这些细胞系可进一步通过在裸鼠体内移植瘤来构建动物模型,为miR-126相关通路的研究及药物开发等的体内和体外实验构建模型提供良好工具。本发明通过对靶标软件设计优选一定数量的miR-126基因上游和下游CRISPR-Cas9靶标序列,分别设计合成针对上游和下游靶标序列的sgRNA单链,并构建到质粒载体中,通过转染293T细胞株,sgRNA与trRNA(trans-activatingRNA)构成特异的识别结构,从而引导Cas9酶特异地剪切miR-126基因两端的对应序列,持续药筛,得到miR-126全长基因敲除的细胞株。通过提取细胞DNA,PCR扩增,并运用DNA水平基因敲除检测的金标准技术——一代测序技术进行确定和验证,得到miR-126基因敲除CRISPR-Cas9的两组有效的上下游sgRNA组合,荧光定量PCR技术显示该组合实现miR-126分子敲除效率高达90%以上,以此构建应用试剂盒,可以对293T、肺癌细胞系A549、乳腺癌细胞系T47D以及血管内皮细胞HUVEC系等多种类型的细胞系进行稳定准确地特异性的全长miR-126基因序列敲除。为解决上述技术问题,本发明采用如下技术方案:在本发明的一方面,提供一种基于CRISPR-Cas9技术的miR-126全长基因敲除试剂盒,该试剂盒包括由质粒pSpCas9(BB)-2A-Puro-sgRNA1和质粒pSpCas9(BB)-2A-Puro-sgRNA3组成的sgRNA质粒组合;所述质粒pSpCas9(BB)-2A-Puro-sgRNA1,含有一段互补的DNA序列,该DNA序列能被转录为特异识别序列如SEQIDNO.2所示的miR-126全长基因的靶标序列的sgRNA,该sgRNA能与trRNA构成特异的识别结构,以致引导Cas9酶特异地剪切miR-126基因对应序列;所述质粒pSpCas9(BB)-2A-Puro-sgRNA3,含有一段互补的DNA序列,该DNA序列能被转录为特异识别序列如SEQIDNO.4所示的miR-126全长基因的靶标序列的sgRNA,该sgRNA能与trRNA构成特异的识别结构,以致引导Cas9酶特异地剪切miR-126基因对应序列;或者,该试剂盒包括由质粒pSpCas9(BB)-2A-Puro-sgRNA2和质粒pSpCas9(BB)-2A-Puro-sgRNA5组成的sgRNA质粒组合;所述质粒pSpCas9(BB)-2A-Puro-sgRNA2,含有一段互补的DNA序列,该DNA序列能被转录为特异识别序列如SEQIDNO.3所示的miR-126全长基因的靶标序列的sgRNA,该sgRNA能与trRNA构成特异的识别结构,以致引导Cas9酶特异地剪切miR-126基因对应序列;所述质粒pSpCas9(BB)-2A-Puro-sgRNA5,含有一段互补的DNA序列,该DNA序列能被转录为特异识别序列如SEQIDNO.6所示的miR-126全长基因的靶标序列的sgRNA,该sgRNA能与trRNA构成特异的识别结构,以致引导Cas9酶特异地剪切miR-126基因对应序列。作为本发明优选的技术方案,所述质粒pSpCas9(BB)-2A-Puro-sgRNA1含有的互补DNA序列为SEQIDNO.7和SEQIDNO.8。作为本发明优选的技术方案,所述质粒pSpCas9(BB)-2A-Puro-sgRNA3含有的互补DNA序列为SEQIDNO.11和SEQIDNO.12。作为本发明优选的技术方案,所述质粒pSpCas9(BB)-2A-Puro-sgRNA2含有的互补DNA序列为SEQIDNO.9和SEQIDNO.10。作为本发明优选的技术方案,所述质粒pSpCas9(BB)-2A-Puro-sgRNA5含有的互补DNA序列为SEQIDNO.15和SEQIDNO.16。作为本发明优选的技术方案,所述质粒pSpCas9(BB)-2A-Puro-sgRNA1和质粒pSpCas9(BB)-2A-Puro-sgRNA3组成的sgRNA质粒组合,所述质粒pSpCas9(BB)-2A-Puro-sgRNA2和质粒pSpCas9(BB)-2A-Puro-sgRNA5组成的sgRNA质粒组合是用包括如下步骤的方法构建的:步骤一,通过对靶标软件设计优选一定数量的miR-126基因上游和下游CRISPR-Cas9靶标序列,具体如下:上游:sgRNA-1:TAATGTCCCGTCGCCAGCGG,如SEQIDNO.2所示;sgRNA-2:GCCACGCCTCCGCTGGCGAC,如SEQIDNO.3所示;下游:sgRNA-3:TCTCAGCGGCGTTTTCGATG,如SEQIDNO.4所示;sgRNA-4:GAGTAATAATGCGCCGTCCA,如SEQIDNO.5所示;sgRNA-5:TTTCGATGCGGTGCCGTGGA,如SEQIDNO.6所示;步骤二,分别设计合成针对上游和下游靶标序列的sgRNA片段,并构建到质粒载体中;所述sgRNA片段序列如下:sgRNA-1F:ccacgTAATGTCCCGTCGCCAGCGG,如SEQIDNO.7所示;sgRNA-1R:aaacCCGCTGGCGACGGGACATTAc,如SEQIDNO.8所示;sgRNA-2F:ccacGCCACGCCTCCGCTGGCGAC,如SEQIDNO.9所示;sgRNA-2R:aaacGTCGCCAGCGGAGGCGTGGC,如SEQIDNO.10所示;sgRNA-3FccacgTCTCAGCGGCGTTTTCGATG,如SEQIDNO.11所示;sgRNA-3RaaacCATCGAAAACGCCGCTGAGAC,如SEQIDNO.12所示;sgRNA-4FccacGAGTAATAATGCGCCGTCCA,如SEQIDNO.13所示;sgRNA-4RaaacTGGACGGCGCATTATTACTC,如SEQIDNO.14所示;sgRNA-5FccacgTTTCGATGCGGTGCCGTGGA,如SEQIDNO.15所示;sgRNA-5RaaacTCCACGGCACCGCATCGAAAc,如SEQIDNO.16所示;步骤三,通过转染293T细胞株,sgRNA与trRNA构成特异的识别结构,从而引导Cas9酶特异地剪切miR-126基因两端的对应序列,持续药筛,得到miR-126全长基因敲除的细胞株;步骤四,通过提取细胞DNA,PCR扩增,测序进行确定和验证,得到优化的两组miR-126基因敲除CRISPR-Cas9的sgRNA质粒组合,即所述质粒pSpCas9(BB)-2A-Puro-sgRNA1和质粒pSpCas9(BB)-2A-Puro-sgRNA3组成的sgRNA质粒组合,以及所述质粒pSpCas9(BB)-2A-Puro-sgRNA2和质粒pSpCas9(BB)-2A-Puro-sgRNA5组成的sgRNA质粒组合。作为本发明优选的技术方案,步骤二中,所述构建到质粒载体中,具体包括如下步骤:(1)以pSpCas9(BB)-2A-Puro质粒为起始载体,用BbsI进行单酶切,对线性化载体进行纯化;(2)将合成好的sgRNA片段稀释并退火;(3)利用T4DNA连接酶将线性化的pSpCas9(BB)-2A-Puro同退火后的sgRNA片段相连,将全部连接产物加入大肠杆菌DH5α感受态细胞转化;(4)利用菌落PCR法对转化后DH5α感受态细胞菌液涂布的平板长出的单菌落进行鉴定,获得表达质粒。在本发明的另一方面,提供上述试剂盒在制备细胞系miR-126基因敲除产品中的应用。所述细胞系包括工具细胞系293T,肿瘤细胞系(例如,肺癌细胞系A549、乳腺癌细胞系T47D)以及内皮细胞HUVEC系。与现有技术相比,本发明具有如下有益效果:本发明基于CRISPR-Cas9技术的miR-126全长基因敲除试剂盒,主要采用CRISPR/Cas9系统敲除miR-126目的基因。这是首次利用CRISPR/Cas9系统特异性的将miR-126全长基因序列进行敲除。本发明具有如下优点:1.克服传统封闭序列方法研究microRNA功能的弊端,miR-126敲除结果可量化监测,miR-126分子敲除效率高达90%以上;2.克服了TALEN技术的耗时繁琐的缺陷,加速了细胞筛选试验并缩减至1周,稳定构建细胞株实验至2个月;构建步骤和实验操作简单,同时降低了试剂成本消耗;3.在CRISPR-Cas9技术上做了技术延伸设计,在miR-126基因序列的上下游两端都设计sgRNA,进行两个位点的同时定点剪切,在DNA水平上实现了对整个miR-126基因序列完整敲除(见图15),一代测序结果显示,可在多种细胞中重复实验效果,稳定性好。本发明极大地避免采用传统的针对单个位点设计sgRNA引导Cas9靶标序列上进行定向剪切方法所导致非同源末端连接途径NHEJ引入随机突变发生的可能性,可以精确地实现microRNA全长编码序列在基因组水平上完整敲除。附图说明图1是本发明实施例1中PCR法验证pSpCas9(BB)-2A-Puro-sgRNA载体构建示意图;其中,通道1-5为pSpCas9(BB)-2A-Puro-sgRNA载体构建阳性克隆PCR结果;通道M为DNA分子量标准。图2是本发明实施例2中pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3配对sgRNA组合在293T中敲除结果示意图;其中,通道3,4,5为pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3组合敲除PCR阳性结果;通道1和2为pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA5组合敲除PCR阴性结果,通道6为对照293T细胞PCR结果;通道M为DNA分子量标准。图3是本发明实施例2中pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3配对sgRNA组合在293T中敲除验证测序结果示意图。图4是本发明实施例2中pSpCas9(BB)-2A-Puro-sgRNA2和pSpCas9(BB)-2A-Puro-sgRNA5配对sgRNA组合在293T中敲除结果示意图;其中,通道1为pSpCas9(BB)-2A-Puro-sgRNA2和pSpCas9(BB)-2A-Puro-sgRNA5组合敲除PCR的阳性结果,通道2,3,4,5,6,7,8为pSpCas9(BB)-2A-Puro-sgRNA2和pSpCas9(BB)-2A-Puro-sgRNA4组合敲除PCR阴性结果,通道9为对照293T细胞PCR结果;通道M为DNA分子量标准。图5是本发明实施例2中pSpCas9(BB)-2A-Puro-sgRNA2和pSpCas9(BB)-2A-Puro-sgRNA5配对sgRNA组合在293T中敲除验证测序结果示意图。图6是本发明实施例2中转染后293T经药筛分离得到的单克隆细胞PCR验证敲除结果示意图;其中,通道1为293T敲除miR-126单克隆细胞的PCR阳性结果,通道2为对照293T细胞PCR未敲除结果;通道M为DNA分子量标准。图7是本发明实施例2中转染后293T经药筛分离得到的敲除阳性的单克隆细胞PCR片段测序结果示意图。图8是本发明实施例2中miR-126敲除293T稳定株荧光定量qPCR检测结果示意图;其中,293Tblank为293T对照细胞未敲除miR-126的荧光定量qPCR结果,293TcloneD2为293T敲除miR-126阳性克隆D2细胞的荧光定量qPCR结果。图9是本发明实施例3中转染后A549经药筛分离得到的单克隆细胞PCR验证敲除结果示意图;其中,通道1,2,3,4,5,6均为A549敲除miR-126后PCR阳性结果,通道7为对照A549细胞未经miR-126敲除的PCR结果;通道M为DNA分子量标准。图10是本发明实施例3中转染后A549经药筛分离得到的敲除阳性的单克隆细胞PCR片段测序结果示意图。图11是本发明实施例4中转染后HUVEC经药筛分离得到的单克隆细胞PCR验证敲除结果示意图;其中,通道2为HUVEC敲除miR-126的阳性PCR结果,通道1,3,4为对照HUVEC细胞miR-126未敲除的PCR结果;通道M为DNA分子量标准。图12是本发明实施例4中转染后HUVEC经药筛分离得到的敲除miR-126阳性的单克隆细胞PCR片段测序结果示意图。图13是本发明实施例5中转染后T47D经药筛后细胞PCR验证敲除结果示意图;其中,通道1,2为T47D细胞样本敲除miR-126的阳性PCR结果,通道3,4为对照T47D细胞miR-126未敲除的PCR结果;通道M为DNA分子量标准。图14是本发明实施例5中转染后T47D经药筛后miR-126敲除阳性的细胞PCR片段测序结果示意图。图15是miR-126基因序列完整敲除示意图。具体实施方式下面结合具体实施例进一步阐述此发明。应理解的是,在此描述的特定实施方式通过举例的方式来表示,并不作为对本发明的限制。在不偏离本发明范围的情况下,本发明的主要特征可以用于各种实施方式。实施例1.载体构建(1)miR-126靶标设计针对miR-126基因(基因名miR-126,基因ID号:406913,基因详细信息见http://www.ncbi.nlm.nih.gov/gene/406913),从该网站中下载得到miR-126基因组序列:5’-CGCTGGCGACGGGACATTATTACTTTTGGTACGCGCTGTGACACTTCAAACTCGTACCGTGAGTAATAATGCGCCGTCCACGGCA-3’,如SEQIDNO.1所示。利用在线软件FengZhanglab'sTargetFinder(http://crispr.mit.edu/)设计sgRNA,输入miR-126基因组序列和其上下游各50bp的序列,设置并检索获得若干sgRNA序列,通过分析sgRNA在基因序列上的位置以及该sgRNA的off-target(脱靶)信息,从中分别选择最优的上游2个靶序列,以及下游3个靶序列,具体如下:上游:sgRNA-1:TAATGTCCCGTCGCCAGCGG,如SEQIDNO.2所示;sgRNA-2:GCCACGCCTCCGCTGGCGAC,如SEQIDNO.3所示;下游:sgRNA-3:TCTCAGCGGCGTTTTCGATG,如SEQIDNO.4所示;sgRNA-4:GAGTAATAATGCGCCGTCCA,如SEQIDNO.5所示;sgRNA-5:TTTCGATGCGGTGCCGTGGA,如SEQIDNO.6所示;(2)合成sgRNA片段根据sgRNA拟克隆进的载体pSpCas9(BB)-2A-Puro(购自于Addgene)的酶切位点BbsI的信息,将上述sgRNA序列获得互补序列后,再加上BbsI限制性内切酶相应的粘性末端。得到如下序列:sgRNA-1F:ccacgTAATGTCCCGTCGCCAGCGG,如SEQIDNO.7所示;sgRNA-1R:aaacCCGCTGGCGACGGGACATTAc,如SEQIDNO.8所示;sgRNA-2F:ccacGCCACGCCTCCGCTGGCGAC,如SEQIDNO.9所示;sgRNA-2R:aaacGTCGCCAGCGGAGGCGTGGC,如SEQIDNO.10所示;sgRNA-3FccacgTCTCAGCGGCGTTTTCGATG,如SEQIDNO.11所示;sgRNA-3RaaacCATCGAAAACGCCGCTGAGAC,如SEQIDNO.12所示;sgRNA-4FccacGAGTAATAATGCGCCGTCCA,如SEQIDNO.13所示;sgRNA-4RaaacTGGACGGCGCATTATTACTC,如SEQIDNO.14所示;sgRNA-5FccacgTTTCGATGCGGTGCCGTGGA,如SEQIDNO.15所示;sgRNA-5RaaacTCCACGGCACCGCATCGAAAc,如SEQIDNO.16所示。将上述序列,送Thermofisher公司合成单链核苷酸链。(3)sgRNA片段同表达载体连接并转化将1μgpSpCas9(BB)-2A-Puro用BbsI(购自于NEB公司,货号R0539S)进行单酶切,37℃水浴1小时后,利用PCR产物纯化试剂盒(购自于Tiagen公司,货号为DP204)对线性化载体进行纯化。将合成好的每组sgRNA的F和R链的干粉稀释至100μM,各取1μL溶液混合于PCR管内,在PCR仪中以每分钟1.5℃逐渐从95℃降至22℃。利用T4DNA连接酶(购自Takara公司,货号6022Q)将1μL线性化的pSpCas9(BB)-2A-Puro同0.5μL退火后的sgRNA片段相连,连接体系见表1:表1sgRNA0.5μL线性化的pSpCas9(BB)-2A-Puro1μLDNAsolutionI5μLddH2Oupto10μL16℃连接15分钟后,将全部连接产物加入至100μL大肠杆菌DH5α感受态细胞(pfu≥108)中,冰上放置30分钟后,于42℃水浴热激90秒,再置于冰上5分钟,于超净台中加入1mLLB液体培养基(Luria-Bertani培养基),37℃振荡培养1小时,取500μL涂布于含Amp(Ampicillin,氨苄西林)的LB固体培养基上,待液体吸收完毕后,倒置于37℃培养过夜。(4)转化后鉴定利用菌落PCR法对转化后DH5α感受态细胞菌液涂布的平板长出的单菌落进行鉴定。菌落PCR鉴定用引物如下:PrimerF:5'GACTATCATATGCTTACCG3',如SEQIDNO.17所示;PrimerR:5'CCAAGTGGGCAGTTTACC3',如SEQIDNO.18所示;菌液PCR体系如表2所示:表2成分体积10×buffer2.5μLprimerF(10μM)0.2μLprimerR(10μM)0.2μLdNTPs(2.5mM)2μLrTaq酶(50U/μL)0.5μL菌落沾取少许ddH2Oupto25μLPCR程序为95℃10min;95℃20sec,55℃30sec,72℃30sec,共35个循环;72℃10min。将得到的PCR产物经1%琼脂糖凝胶电泳去检测,出现100bp左右扩增片段的单菌落为含有阳性克隆的单菌落(如图1所示,PCR产物片段大小同DNA分子量标准的100bp片段大小相近,而1000bp片段为该分子量标准的最亮片段)。将阳性克隆单菌落接种2mLLB液体培养基中,37℃过夜振荡培养后取1mL菌液用于保种,剩余1mL菌液送测序公司(上海美吉生物医药科技有限公司)进行测序验证。将得到的测序结果经Blast分析表明,sgRNA片段已克隆进pSpCas9(BB)-2A-Puro质粒中,且序列一致,可以用于后续实验。带有sgRNA的载体分别命名为pSpCas9(BB)-2A-Puro-sgRNA1,pSpCas9(BB)-2A-Puro-sgRNA2,pSpCas9(BB)-2A-Puro-sgRNA3,pSpCas9(BB)-2A-Puro-sgRNA4以及pSpCas9(BB)-2A-Puro-sgRNA5。实施例2.质粒转染和293T细胞中miR-126敲除活性鉴定以及293T敲除miR-126细胞模型的构建(1)质粒抽提将实施例1中经过测序验证的,分别包含sgRNA-1、sgRNA-2、sgRNA-3、sgRNA-4、sgRNA-5片段的pSpCas9(BB)-2A-Puro质粒的大肠杆菌DH5α各自接种于5mLLB液体培养基中,37℃过夜振荡培养。利用无内毒素小量质粒提取试剂盒(试剂盒为Omega公司出品,货号为D6948-02)对这些菌液进行质粒抽提。抽提后的质粒再在超净台中无水乙醇沉淀后,用无菌水溶解进行对质粒的无菌处理。(2)293T细胞转染一对含sgRNA的pSpCas9(BB)-2A-Puro-sgRNA质粒1)293T细胞培养将293T细胞以3×105细胞/孔的密度铺于24孔板中,确保各孔细胞生长状态良好,密度相仿,待细胞为单层并处于对数中期,细胞汇合度达80%左右进行转染。2)制备转染复合物:根据表3的配对,将一对含sgRNA的pSpCas9(BB)-2A-Puro-sgRNA质粒通过Lipofectamine2000脂质体试剂转染293T细胞。表3上下游含sgRNA质粒配对表序号上游sgRNA质粒下游sgRNA质粒1pSpCas9(BB)-2A-Puro-sgRNA1pSpCas9(BB)-2A-Puro-sgRNA32pSpCas9(BB)-2A-Puro-sgRNA1pSpCas9(BB)-2A-Puro-sgRNA53pSpCas9(BB)-2A-Puro-sgRNA2pSpCas9(BB)-2A-Puro-sgRNA44pSpCas9(BB)-2A-Puro-sgRNA2pSpCas9(BB)-2A-Puro-sgRNA5取2个无菌EP管,分别在50μL的无血清无抗生素的Opti-MEM培养基中稀释质粒0.8μg和2μL的Lipofectamine2000,室温静置5分钟,将稀释的质粒和Lipofectamine2000混合,共得到100μL的转染复合物。轻轻混匀后,室温孵育20分钟。3)细胞转染:将上述转染复合物加入到对应的24孔板中,来回轻柔摇晃细胞培养板,将细胞放回37℃5%CO2培养箱继续培养6小时后,将培养基更换为完全培养基,继续将细胞置于37℃5%CO2培养箱培养。4)药筛转染24小时后,将培养基更换为含2μg/mLpuromycin的完全培养基。每24小时更换一次含药物的完全培养基,连续三天。每天在显微镜下观察绿色荧光信号,记录下细胞状态。(3)CRISPR/Cas9敲除效率验证1)PCR测序方法验证基因组水平上的敲除效率收集药筛后的293T细胞以及未经转染的293T细胞,利用超微量样品基因型鉴定试剂盒(南京尧顺禹生物科技有限公司,KC-101)进行样本处理,并利用如表4所示PCR引物进行PCR扩增鉴定。表4*引物序列(5'--3')*引物名称GAGGGAGGATAGGTGGGTTC,如SEQIDNO.19所示mir126-test-FwAGGCAGAGCCAGAAGACTCA,如SEQIDNO.20所示mir126-test-RvGCACTGGAATCTGGGCGGAAGG,如SEQIDNO.21所示mir126-test-2-FwAGAGCCAGGCGCTGGGTCAC,如SEQIDNO.22所示mir126-test-2-RvPCR反应体系如表5所示:表594℃5min;94℃30sec,62℃30sec,72℃30sec,共35个循环;72℃10min。将PCR扩增产物稀释10倍,以稀释物为模板进行第二轮PCR,PCR体系如表6所示:表694℃5min;94℃30sec,62℃30sec,72℃2min,共35个循环;72℃5min。将PCR扩增产物经1%琼脂糖凝胶电泳检测(结果见图2,图4所示),选取同未经转染的293T细胞样本条带相比明显偏小的PCR产物送去测序(上海美吉生物医药科技有限公司)(结果见图3,图5所示)。测序结果利用Blast进行分析表明,由pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3以及由pSpCas9(BB)-2A-Puro-sgRNA2和pSpCas9(BB)-2A-Puro-sgRNA5组成的sgRNA质粒组合可完全敲除293T细胞中miR-126基因组序列,且pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3组合删除能得到阳性结果的数量更多,因此效率更佳。其他sgRNA配对组合敲除验证结果为阴性,不能有效敲除293T细胞中miR-126基因组序列。由此,选定pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3,pSpCas9(BB)-2A-Puro-sgRNA2和pSpCas9(BB)-2A-Puro-sgRNA5为miR-126全长基因敲除试剂盒的sgRNA组合;优选pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3组合。(4)构建miR-126敲除293T细胞模型并qPCR测试敲除效率由选定的pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3为miR-126全长基因敲除试剂盒的sgRNA组合,按照实施例2的步骤(1)——(3)得到含miR-126敲除的293T细胞系,将已验证含miR-126敲除的293T细胞计数后,稀释至2个/孔的数量接种24孔板,置于37℃,5%CO2培养箱培养两周,期间每天观察孔板中单克隆的生长状态。两周后,如孔板中单克隆生长成细胞团,取细胞团中细胞进行PCR验证测序,看该细胞团是否为敲除miR-126的293T单克隆细胞团(结果详见图6所示),PCR结果显示293T单克隆细胞团的条带明显小于对照293T细胞的PCR条带,将293T单克隆细胞团的PCR产物送测序(上海美吉生物医药科技有限公司,结果如图7所示),测序结果经Blast比对验证,miR-126基因组编码序列在293T单克隆细胞团(单克隆命名为D2)中完全被敲除,将验证正确的单克隆细胞团D2扩大培养,至此miR-126敲除的293T细胞模型构建成功。将构建成功的293T敲除miR-126细胞稳定株克隆D2扩大培养并利用QIAGENmiRNeasyMiniKit(QIAGEN,货号217004)进行RNA(包含microRNA)抽提,抽提后的RNA利用miScriptIIRTKit(QIAGEN,货号218160)进行反转,体系如表7:表737℃60min;95℃5min将反转好的cDNA稀释10倍后,利用如表8所示引物进行qPCR。表8*引物序列(5'--3')*引物名称TCGTACCGTGAGTAATAATGCG,如SEQIDNO.23所示mir126FwGATTGAATCGAGCACCAGTTAC,如SEQIDNO.24所示commonqTTCGTGAAGCGTTCCATATTTT,如SEQIDNO.25所示U6FwPCR反应体系如表9:表995℃2min;94℃15sec,60℃1min共40个循环;60℃1min时荧光定量PCR仪SYBR通道采集信号。实验结束后,利用荧光定量PCR仪自带软件分析实验结果,获得293T单克隆D2(已验证敲除成功的239T单克隆稳定株)的miR-126相对于自身U6的表达量,以及空白对照样本的miR-126相对于自身U6的表达量。以空白对照样本的miR-126相对表达量为标准,将其定义为表达效率为100%,从而计算出293T单克隆D2中miR-126的表达效率为5.37%(结果如图8所示),表明利用pSpCas9(BB)-2A-Puro-sgRNA1和pSpCas9(BB)-2A-Puro-sgRNA3sgRNA组合可敲除293T中miR-126,且敲除效率可达94.63%。实施例3构建miR-126敲除的肺癌细胞模型1)肺癌细胞A549细胞转染将pSpCas9(BB)-2A-Puro-sgRNA1、pSpCas9(BB)-2A-Puro-sgRNA3利用Lipofectamine2000共转染肺癌细胞A549,转染24小时后,利用含2μg/mLpuromycin的完全培养基进行药筛,药筛三天后,收集细胞。2)敲除活性验证收集药筛后的A549细胞以及未经转染的A549细胞,利用超微量样品基因型鉴定试剂盒(南京尧顺禹生物科技有限公司,KC-101)进行样本处理,并利用如表10所示PCR引物进行PCR扩增鉴定。表10*引物序列(5'--3')*引物名称GAGGGAGGATAGGTGGGTTC,如SEQIDNO.19所示mir126-test-FwAGGCAGAGCCAGAAGACTCA,如SEQIDNO.20所示mir126-test-RvGCACTGGAATCTGGGCGGAAGG,如SEQIDNO.21所示mir126-test-2-FwAGAGCCAGGCGCTGGGTCAC,如SEQIDNO.22所示mir126-test-2-RvPCR反应体系如表11:表1194℃5min;94℃30sec,62℃30sec,72℃30sec,共35个循环;72℃10min。将PCR扩增产物稀释10倍,以稀释物为模板进行第二轮PCR,PCR体系如表12:表1294℃5min;94℃30sec,62℃30sec,72℃2min,共35个循环;72℃5min。将PCR扩增产物经1%琼脂糖凝胶电泳检测,选取同未经转染的A549细胞样本条带相比明显偏小的PCR产物送去测序(上海美吉生物医药科技有限公司)。测序结果利用Blast进行分析表明,miR-126片段在基因组中被有效敲除。3)分离miR-126敲除的肺癌细胞系A549单克隆将已验证含miR-126敲除的肺癌细胞系A549细胞计数后,稀释至2个/孔的数量接种24孔板,置于37℃,5%CO2培养箱培养两周,期间每天观察孔板中单克隆的生长状态。两周后,如孔板中单克隆生长成细胞团,取细胞团中细胞进行PCR验证测序,看该细胞团是否为敲除miR-126的A549单克隆细胞团(结果详见图9所示),PCR结果显示A549的6个单克隆条带均明显小于对照A549细胞的PCR条带,随机挑选6个单克隆中单克隆A2的PCR产物送测序(上海美吉生物医药科技有限公司,结果如图10所示),测序结果经Blast比对验证,miR-126在A549单克隆A2中完全被敲除,将验证正确的单克隆细胞团培养扩大培养,至此miR-126敲除的A549细胞模型构建成功。实施例4构建miR-126敲除的内皮细胞模型1)人脐静脉内皮细胞HUVEC细胞转染将pSpCas9(BB)-2A-Puro-sgRNA1、pSpCas9(BB)-2A-Puro-sgRNA3利用Lipofectamine2000共转染人脐静脉内皮细胞HUVEC,转染24小时后,利用含2μg/mLpuromycin的完全培养基进行药筛,药筛三天后,收集细胞。2)敲除活性验证收集药筛后的HUVEC细胞以及未经转染的HUVEC细胞,利用超微量样品基因型鉴定试剂盒(南京尧顺禹生物科技有限公司,KC-101)进行样本处理,并利用如表13所示PCR引物进行PCR扩增鉴定。表13*引物序列(5'--3')*引物名称GAGGGAGGATAGGTGGGTTC,如SEQIDNO.19所示mir126-test-FwAGGCAGAGCCAGAAGACTCA,如SEQIDNO.20所示mir126-test-RvGCACTGGAATCTGGGCGGAAGG,如SEQIDNO.21所示mir126-test-2-FwAGAGCCAGGCGCTGGGTCAC,如SEQIDNO.22所示mir126-test-2-RvPCR反应体系如表14:表1494℃5min;94℃30sec,62℃30sec,72℃30sec,共35个循环;72℃10min。将PCR扩增产物稀释10倍,以稀释物为模板进行第二轮PCR,PCR体系如表15:表1594℃5min;94℃30sec,62℃30sec,72℃2min,共35个循环;72℃5min。将PCR扩增产物经1%琼脂糖凝胶电泳检测,选取同未经转染的HUVEC细胞样本条带相比明显偏小的PCR产物送去测序(上海美吉生物医药科技有限公司)。测序结果利用Blast进行分析表明,miR-126片段在基因组中被有效敲除。3)分离miR-126敲除的HUVEC单克隆将已验证含miR-126敲除的HUVEC细胞计数后,稀释至2个/孔的数量接种24孔板,置于37℃,5%CO2培养箱培养两周,期间每天观察孔板中单克隆的生长状态。两周后,如孔板中单克隆生长成细胞团,取细胞团中细胞进行PCR验证测序,看该细胞团是否为敲除miR-126的HUVEC单克隆细胞团(结果详见图11所示),PCR结果显示HUVEC的单克隆细胞团条带明显小于对照HUVEC细胞的PCR条带,将HUVEC单克隆细胞团(命名为单克隆H1)的PCR产物送测序(上海美吉生物医药科技有限公司,结果如图12所示),测序结果经Blast比对验证,miR-126在HUVEC单克隆细胞团H1中完全被敲除,将验证正确的单克隆细胞团培养扩大培养,至此miR-126敲除的HUVEC细胞模型构建成功。实施例5乳腺癌细胞T47D细胞系miR-126敲除1)乳腺癌细胞T47D细胞转染将pSpCas9(BB)-2A-Puro-sgRNA1、pSpCas9(BB)-2A-Puro-sgRNA3利用Lipofectamine2000共转染乳腺癌细胞T47D,转染24小时后,利用含2μg/mLpuromycin的完全培养基进行药筛,药筛三天后,收集细胞。2)敲除活性验证收集药筛后的T47D细胞以及未经转染的T47D细胞,利用超微量样品基因型鉴定试剂盒(南京尧顺禹生物科技有限公司,KC-101)进行样本处理,并利用如表16所示的PCR引物进行PCR扩增鉴定。表16*引物序列(5'--3')*引物名称GAGGGAGGATAGGTGGGTTC,如SEQIDNO.19所示mir126-test-FwAGGCAGAGCCAGAAGACTCA,如SEQIDNO.20所示mir126-test-RvGCACTGGAATCTGGGCGGAAGG,如SEQIDNO.21所示mir126-test-2-FwAGAGCCAGGCGCTGGGTCAC,如SEQIDNO.22所示mir126-test-2-RvPCR反应体系如表17:表1794℃5min;94℃30sec,62℃30sec,72℃30sec,共35个循环;72℃10min。将PCR扩增产物稀释10倍,以稀释物为模板进行第二轮PCR,PCR体系如表18:表1894℃5min;94℃30sec,62℃30sec,72℃2min,共35个循环;72℃5min。将PCR扩增产物经1%琼脂糖凝胶电泳检测,选取同未经转染的T47D细胞样本条带相比明显偏小的PCR产物(如图13所示)送去测序(上海美吉生物医药科技有限公司)。测序结果(如图14所示)利用Blast进行分析表明,miR-126片段在基因组中被有效敲除。依据上述实验结果,本发明确定针对miR-126基因设计的sgRNA-1和sgRNA-3;sgRNA-2和sgRNA-5为有效sgRNA序列组合,其中包含sgRNA-1、sgRNA3质粒组合可作为miR-126全长基因敲除的试剂盒的Cas9质粒。实验证明,该试剂盒可有效实现细胞水平的基因敲除,并可用于多种细胞系包括工具细胞系293T,肿瘤细胞系:肺癌细胞系A549、乳腺癌细胞系T47D以及内皮细胞HUVEC系等细胞系miR-126的基因敲除。该miR-126基因敲除的各种细胞模型,可用于血管内皮细胞分化不良,以及多种恶性肿瘤如:乳腺癌、肺癌等方面的基因功能研究,为后续动物建模奠定了基础,为进一步药物开发等应用研究提供了便利的工具。序列表<110>上海伯豪生物技术有限公司<120>一种基于CRISPR-Cas9技术的miR-126全长基因敲除试剂盒及其应用<130>HJ16-12192<160>25<170>PatentInversion3.5<210>1<211>85<212>DNA<213>人工序列<400>1cgctggcgacgggacattattacttttggtacgcgctgtgacacttcaaactcgtaccgt60gagtaataatgcgccgtccacggca85<210>2<211>20<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(20)<223>sgRNA<400>2taatgtcccgtcgccagcgg20<210>3<211>20<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(20)<223>sgRNA<400>3gccacgcctccgctggcgac20<210>4<211>20<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(20)<223>sgRNA<400>4tctcagcggcgttttcgatg20<210>5<211>20<212>sgRNA<213>人工序列<220><221>misc_RNA<222>(1)..(20)<223>sgRNA<400>5gagtaataatgcgccgtcca20<210>6<211>20<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(20)<223>sgRNA<400>6tttcgatgcggtgccgtgga20<210>7<211>25<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(25)<223>sgRNA<400>7ccacgtaatgtcccgtcgccagcgg25<210>8<211>25<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(25)<223>sgRNA<400>8aaacccgctggcgacgggacattac25<210>9<211>24<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(24)<223>sgRNA<400>9ccacgccacgcctccgctggcgac24<210>10<211>24<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(24)<223>sgRNA<400>10aaacgtcgccagcggaggcgtggc24<210>11<211>25<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(25)<223>sgRNA<400>11ccacgtctcagcggcgttttcgatg25<210>12<211>25<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(25)<223>sgRNA<400>12aaaccatcgaaaacgccgctgagac25<210>13<211>24<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(24)<223>sgRNA<400>13ccacgagtaataatgcgccgtcca24<210>14<211>24<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(24)<223>sgRNA<400>14aaactggacggcgcattattactc24<210>15<211>25<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(25)<223>sgRNA<400>15ccacgtttcgatgcggtgccgtgga25<210>16<211>25<212>RNA<213>人工序列<220><221>misc_RNA<222>(1)..(25)<223>sgRNA<400>16aaactccacggcaccgcatcgaaac25<210>17<211>19<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>17gactatcatatgcttaccg19<210>18<211>18<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>18ccaagtgggcagtttacc18<210>19<211>20<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>19gagggaggataggtgggttc20<210>20<211>20<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>20aggcagagccagaagactca20<210>21<211>22<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>21gcactggaatctgggcggaagg22<210>22<211>20<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>22agagccaggcgctgggtcac20<210>23<211>22<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>23tcgtaccgtgagtaataatgcg22<210>24<211>22<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>24gattgaatcgagcaccagttac22<210>25<211>22<212>DNA<213>人工序列<220><221>misc_feature<223>引物<400>25ttcgtgaagcgttccatatttt22当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1