一种医药中间体2,6-二氟-3-吗啉苯基甲胺及其制备方法与流程
本发明涉及一种医药中间体2,6-二氟-3-吗啉苯基甲胺及其制备方法,即如式1所示结构化合物及制备方法。
背景技术:
多取代苯甲胺类化合物由于其中的氨基可以作为酰胺化、还原氨化等反应的位点,因此在制药工业中有着重要的应用。有研究表明,在多取代苯甲胺类化合物的苯环中同时引入吗啉和氟取代基,可以很好地同时改善药物的溶解性和代谢动力学性质。例如,抗菌性药物Linezolid3的分子中即含有吗啉和氟取代的苯环结构单元;2005年报道的一种HIV整合酶抑制剂中也含有该结构单元。因此,多氟取代的吗啉苯基甲胺衍生物可作为多种药物的重要合成中间体。目前合成此类化合物的方法大多需要首先在苯环上引入卤素(如Br),然后再通过钯催化的Buchward-Hartwig偶联反应引入吗啉结构单元。例如,最近有专利(PTC Int.Appl.,2012093101)报道了2-氟-3-氯-5-吗啉苯基甲胺的合成方法,反应式如下:
其中关键的第一步反应为吗啉与溴代苯的Buchward-Hartwig偶联反应。此方法需要用到较为昂贵的钯催化剂及膦配体,并且具有产率低和不易放大生产等缺陷。
技术实现要素:
本发明的目的是提供一种如式1所示的可作为医药中间体的化合物I,即2,6-二氟-3-吗啉苯基甲胺,同时提供化合物I的制备方法。
本发明的如式1的示化合物I的制备方法如式2示,即:以2,6-二氟苯腈为起始原料,
经过硝化、硝基还原、吗啉环的形成以及氰基的还原等转化,得到目标产物。
本发明用于制备化合物I的详细方法是:
1)2,6-二氟苯腈的硝化
将5.0~10.0g 2,6-二氟苯腈II溶于15.0~40.0mL浓硫酸中,-10~5℃温度下,加入6~30mL浓硝酸,加完后于20~35℃温度下充分反应,冷却后用二氯甲烷萃取,无水硫酸钠干燥,过滤、减压蒸除溶剂得硝化产物III;
2)硝化产物的还原
将6.0~11.0g上步骤所得硝化产物III溶于50~130mL异丙醇中,依次加入质量浓度为40%的氯化铵水溶液34~90g、6.0~20.0g的铁粉,于80~110℃温度下反应6~10小时,冷却后过滤除去不溶物,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,用柱层析进行纯化得氨基化合物IV;
3)化合物V的合成
将0.8~2.8g上步骤所得氨基化合物IV溶于1.0~4.0mL DMF中,依次加入1.2~4.8mL的双(2-溴乙基)醚、0.8~4.0mL的二乙胺,于160~190℃温度下反应2~4小时,冷却后用二氯甲烷稀释,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析纯化得化合物V;
4)2,6-二氟-3-吗啉苯基甲胺的合成
将0.5~1.6g上步骤所得化合物V溶于6.0~20.0mL乙醇中,装入高压釜,依次加入0.2~0.8g的Raney-Ni、8.0~18.0mL的四氢呋喃、0.5~1.5mL的氨水,然后充入5~10大气压的氢气,于50~80℃温度下反应4~8小时,冷却后过滤除去不溶物,减压蒸除溶剂得2,6-二氟-3-吗啉苯基甲胺I;
上述步骤2)和步骤3)中柱层析用石油醚:乙酸乙酯=10:1~2:1为洗脱剂。
本发明中化合物I的制备方法的优选例为:用化合物Ⅱ制备化合物Ⅲ的过程中使用浓硫酸和浓硝酸作为硝化试剂,反应温度为-10~5℃;用化合物Ⅲ制备化合物Ⅳ过程中用铁粉作为还原剂,异丙醇作为溶剂,反应温度为80~110℃;用化合物Ⅳ制备化合物Ⅴ过程中用DMF作为溶剂,二乙胺作为碱,反应温度为160~190℃;用化合物Ⅴ制备目标化合物Ⅰ过程中用Raney-Ni作为氢化催化剂,氢气压力为5~10大气压,反应温度为50~80℃。
本发明所用方法具有原料来源广泛、价格便宜、制备过程易于控制和操作等优点。所得产物可用于制备HIV整合酶抑制剂及治疗年龄相关性黄斑变性药物。
附图说明
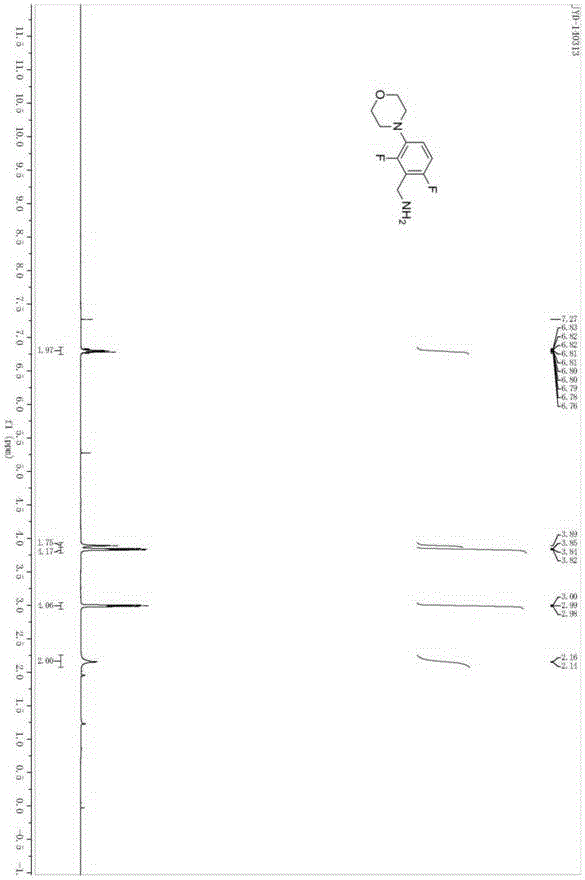
图1是2,6-二氟-3-吗啉苯基甲胺的1HNMR图;
图2是2,6-二氟-3-吗啉苯基甲胺的13CNMR图。
具体实施方式
一种医药中间体2,6-二氟-3-吗啉苯基甲胺的制备方法,包括如下步骤:
将5g 2,6-二氟苯腈II溶于15.0mL浓硫酸中,-10℃温度下加入6.0mL浓硝酸,
加完后于20℃温度下继续反应5小时,冷却后用二氯甲烷萃取,无水硫酸钠干燥,过滤、减压蒸除溶剂得硝化产物III。
2)将6.0g上步骤所得硝化产物III溶于50.0mL异丙醇中,依次加入质量浓度为40%的氯化铵水溶液34g、铁粉(6.0g),于80℃温度下反应10小时,
冷却后过滤除去不溶物,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析(石油醚:乙酸乙酯=6:1~2:1为洗脱剂)纯化得氨基化合物IV。
3)将0.8g上步骤所得氨基化合物IV溶于1.0mL DMF中,依次加入双(2-溴乙基)醚(1.2mL)、二乙胺(0.8mL),于160℃温度下反应4小时,冷却后用
二氯甲烷稀释,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析(石油醚:乙酸乙酯=6:1~2:1为洗脱剂)纯化得化合物V。
4)将0.5g上步骤所得化合物V溶于6.0mL乙醇中,装入高压釜,依次加
入Raney-Ni(0.2g)、四氢呋喃(8.0mL)、氨水(0.5mL),然后充入氢气(5大气压),于50℃温度下反应8小时,冷却后过滤除去不溶物,减压蒸除溶剂得2,6-二氟-3-吗啉苯基甲胺I。四步总收率47%。1HNMR(400MHz,CDCl3):6.83-6.76(m,2H),3.89(s,2H),3.84(t,J=4.0Hz,4H),2.99(t,J=4.0Hz,4H),2.16(s,2H);13CNMR(100MHz,CDCl3):157.6,155.2,152.7,136.6,117.4,110.6,66.9,51.3,34.0.
实施例2:
一种医药中间体2,6-二氟-3-吗啉苯基甲胺的制备方法,包括如下步骤,反应式参见式3至式6:
1)将7.5g 2,6-二氟苯腈II溶于23.0mL浓硫酸中,0℃温度下加入18.0mL浓硝酸,加完后于25℃温度下继续反应3小时,冷却后用二氯甲烷萃取,无水硫酸钠干燥,过滤、减压蒸除溶剂得硝化产物III。
2)将8.0g上步骤所得硝化产物III溶于80.0mL异丙醇中,依次加入质量比为40%的氯化铵水溶液60g、铁粉(13.0g),于90℃温度下反应8小时,冷却后过滤除去不溶物,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析(石油醚:乙酸乙酯=6:1~2:1为洗脱剂)纯化得氨基化合物IV。
3)将1.5g上步骤所得氨基化合物IV溶于2.5mL DMF中,依次加入双(2-溴乙基)醚(2.8mL)、二乙胺(1.8mL),于170℃温度下反应3小时,冷却后用二氯甲烷稀释,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析(石油醚:乙酸乙酯=6:1~2:1为洗脱剂)纯化得化合物V。
4)将1.0g上步骤所得化合物V溶于10.0mL乙醇中,装入高压釜,依次加入Raney-Ni(0.5g)、四氢呋喃(12.0mL)、氨水(0.8mL),然后充入氢气(8大气压),于60℃温度下反应7小时,冷却后过滤除去不溶物,减压蒸除溶剂得2,6-二氟-3-吗啉苯基甲胺I。四步总收率46%。
实施例3:
一种医药中间体2,6-二氟-3-吗啉苯基甲胺的制备方法,包括如下步骤,反应式参见式3至式6:
1)将10.0g 2,6-二氟苯腈II溶于40.0mL浓硫酸中,5℃温度下加入30.0mL浓硝酸,加完后于35℃温度下继续反应2小时,冷却后用二氯甲烷萃取,无水硫酸钠干燥,过滤、减压蒸除溶剂得硝化产物III。
2)将11.0g上步骤所得硝化产物III溶于130.0mL异丙醇中,依次加入质量浓度为40%的氯化铵水溶液90g、铁粉(20.0g),于110℃温度下反应6小时,冷却后过滤除去不溶物,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析(石油醚:乙酸乙酯=6:1~2:1为洗脱剂)纯化得氨基化合物IV。
3)将2.8g上步骤所得氨基化合物IV溶于4.0mL DMF中,依次加入双(2-溴乙基)醚(4.8mL)、二乙胺(4.0mL),于190℃温度下反应2小时,冷却后用二氯甲烷稀释,有机相依次用水、饱和食盐水洗涤,无水硫酸钠干燥,过滤、减压蒸除溶剂,柱层析(石油醚:乙酸乙酯=6:1~2:1为洗脱剂)纯化得化合物V。
4)将1.6g上步骤所得化合物V溶于20.0mL乙醇中,装入高压釜,依次加入Raney-Ni(0.8g)、四氢呋喃(18.0mL)、氨水(1.5mL),然后充入氢气(10大气压),于80℃温度下反应4小时,冷却后过滤除去不溶物,减压蒸除溶剂得2,6-二氟-3-吗啉苯基甲胺I。四步总收率45%。
- 还没有人留言评论。精彩留言会获得点赞!