用于基因编辑的组合物和方法与流程
本申请要求于2015年12月21日提交的中国专利申请no.201510971234.5和2016年5月25日提交的中国专利申请no.201610349444.5的优先权权益,每篇专利申请内容全文以引用方式并入本文中。
提交序列表的ascii文本文件
将以下以ascii文本文件提交的内容全文以引用方式并入本文:序列表的计算机可读形式(crf)(文件名称:777072000140seqlist.txt,记录日期:2016年12月15日,大小:309kb)。
本发明涉及可用于靶核酸的序列特异性修饰(包括细胞中基因编辑)的基于argonaute的方法、组合物和递送系统。
背景技术:
对基因信息的深入了解和基因疗法的开发需要准确而有效的基因编辑工具。作为基因工程中迅速发展的技术领域,基因或基因组编辑采用核酸酶将序列特异性修饰(诸如突变、插入和置换等)引入基因或基因组中。当前的基因编辑技术通常涉及三个步骤。首先,使一种或多种专门设计的引导dna或rna(引导元件)和内切核酸酶(基因切割元件)彼此结合,其中前种元件将后种元件引导至靶基因的特定基因座。第二,内切核酸酶切割靶基因的特定基因座,在靶基因两条链中的每条中诱导切口或间隙,从而产生双链断裂(dsb)。最后,dsb激活细胞中的内源dna修复机制,该机制修复断裂并同时在修复位点引入修饰,诸如突变、插入和置换。大多数真核细胞依赖于两种dsb修复机制:非同源末端连接(nhej)和同源介导的修复(hdr)。nhej利用一系列酶来直接连接dna的两个断裂末端。因此,nhej是一种易错配、低保真性的修复途径,通常在修复期间引入突变。相比之下,hdr需要同源序列充当模板以在dsb处恢复丢失的dna序列。hdr中对同源模板的需要可用于将外源序列引入基因组的靶基因座中。hdr的机制类似于同源重组。然而,因为hdr是基于dsb的修复途径,使用hdr引入外源序列的效率比使用同源重组约高三个数量级。因此,通过修复和修饰诱导的dsb,nejh和hdr能够使用核酸酶进行高效的基因编辑。基因编辑技术对于我们对基因功能、基因工程、以及基因疗法开发的理解具有革命性的影响。
目前,四种主要的核酸酶家族广泛用于基因编辑:锌指核酸酶(zfn)、转录激活因子样效应因子核酸酶(talen)、工程大范围核酸酶/再工程化归巢内切酶、以及crispr/cas9系统。前三个核酸酶家族的应用受到各种局限性的挑战,其包括需要特定靶序列、用于适应不同靶序列的专用设计的核酸酶,并且脱靶效应较高。crispr/cas9是目前最常用的基因编辑系统。细菌来源的酶即cas9可利用引导rna在基因组中pam序列的近侧和上游的任何靶基因座处理论地切割并诱导dsb。然而,因为rna易于形成二次结构,cas9系统在编辑富gc基因中具有较低效率。另外,由于细胞天然地产生大量rna片段,仍然存在cas9结合非特异性rna的可能性,并从而切割基因组中的非靶基因座。还已知cas9对引导rna和靶dna之间的错配具有相对高的耐受性。例如,引导rna与靶dna之间多至5个核苷酸错配并不妨碍cas9切割。因此,cas9系统受其脱靶效应的限制。
argonaute是普遍存在于细菌、植物、古生菌和动物细胞中的核酸结合蛋白的家族。argonaute在rna干扰(rnai)中的作用最为人们所熟知。来自大多数菌种的argonaute蛋白结合非编码寡核苷酸rna,该非编码寡核苷酸rna充当argonaute蛋白的引导rna,从而经由序列互补性识别靶mrna,并随后诱导mrna降解或转录抑制,由此抑制基因翻译。近年来,来自细菌菌种嗜热栖热菌(thermusthermophilus)的argonaute(“ttago”)示出结合单链dna(ssdna)并将ssdna用作降解dna质粒的引导物。该现象表明,来自一些菌种的argonaute是dna引导的dna核酸内切酶。然而,ttago需要高温(>65℃)来实现其内切核酸酶活性,因此不适合作为基因编辑工具。
本文提及的所有出版物、专利、专利申请和公布的专利申请的公开内容据此全文以引用方式并入本文。
技术实现要素:
本专利申请提供了使用argonaute(ago)蛋白和短单链引导dna修饰靶核酸的方法、组合物、递送系统和试剂盒。靶核酸可存在于体外或细胞内。靶核酸的示例性修饰包括但不限于位点特异性切割、引入突变、插入外源序列、序列置换和基因表达改变。
本申请的一个方面提供了一种修饰靶核酸的方法,包括在约10℃至约60℃的温度(诸如约37℃)下使靶核酸与argonaute(ago)蛋白和单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。
在根据上述任一种方法的一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。
在根据上述任一种方法的一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个(诸如3个、2个、1或0个的任一种)错配。
在根据上述任一种方法的一些实施方案中,靶基因座具有至少约60%(诸如至少约60%、65%、70%、75%、80%的任一种或更大)的gc含量。
在根据上述任一种方法的一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。
在根据上述任一种方法的一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌(natronobacteriumgregoryi)、铜绿微囊藻(microcystisaeruginosa)、halogeometricumpallidum、亚洲嗜盐碱杆菌(natrialbaasiatica)、西藏嗜盐碱红菌(natronorubrumtibetense)、隐红碱线菌(natrinemapellirubrum)、伯林盐几何菌(halogeometricumborinquense)、thermococcusbarophilus、嗜热聚球藻(thermosynechococcuselongatus)、嗜冷嗜盐菌(halorubrumlacusprofundi)、微囊藻属(microcystissp.)、聚球藻属(synechococcussp.)、梭菌属(clostridiumbartlettii)、产气荚膜梭菌(clostridiumperfringens)、煎盘梭菌(clostridiumsartagoforme)、梭菌属(clostridiumsp.)、intestinibacterbartlettii、嗜热古菌(ferroglobusplacidus)、盐杆菌属(halobacteriumsp.)、methanocaldococcusfervens、浅黄假单胞菌(pseudomonasluteola)、thermogladiuscellulolyticus、固氮弧菌属物种(aromatoleumaromaticum)、深海超嗜热古菌(thermococcusonnurineus)、坎式甲烷火菌(methanopyruskandleri)、细长聚球藻(synechococcuselongatus)、好热黄无氧芽孢菌(anoxybacillusflavithermus)、微小杆菌属(exiguobacteriumsp.)、鞘丝藻属(lyngbyasp.)、丁酸梭菌(clostridiumbutyricum)、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌(burkholderiagraminis)、死海盐盒菌(haloarculamarismortui)、百脉根中慢生根瘤菌(mesorhizobiumloti)、红细菌目细菌(rhodobacteralesbacterium)和解肝磷酯土地杆菌(pedobacterheparinus)。在一些实施方案中,ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白来源于解肝磷酯土地杆菌。在一些实施方案中,ago蛋白来源于微囊藻属。在一些实施方案中,ago蛋白来源于铜绿微囊藻。
在根据上述任一种方法的一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:2具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:11具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:41具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。
在根据上述任一种方法的一些实施方案中,引导dna在5′端被磷酸化。
在根据上述任一种方法的一些实施方案中,引导dna的长度为约10至约50个核苷酸(nt),诸如约15nt至约35nt、约20nt至约27nt、或约23nt至25nt。
在根据上述任一种方法的一些实施方案中,接触在二价金属离子诸如mg2+的存在下进行。在一些实施方案中,二价金属离子的浓度为至少约0.1mm。
在根据上述任一种方法的一些实施方案中,靶核酸是分离的dna。在一些实施方案中,靶核酸存在于细胞中。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,该方法包括用引导dna和编码ago蛋白的核酸转染细胞。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,该方法包括将包含ago蛋白和引导dna的预形成的复合物递送到细胞中。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,该方法包括用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被非病毒微生物诸如支原体污染。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,ago蛋白包括核定位信号(nls)。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,靶核酸对于细胞是内源性的。在一些实施方案中,靶核酸是基因组dna。在一些实施方案中,靶核酸对于细胞是外源性的。在一些实施方案中,靶核酸是病毒dna。在一些实施方案中,靶核酸被整合进细胞的基因组中。在一些实施方案中,靶核酸未整合进细胞的基因组中。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,细胞是原核细胞。在一些实施方案中,细胞是真核细胞。在一些实施方案中,细胞是哺乳动物细胞,诸如人细胞。在一些实施方案中,细胞是酵母细胞、真菌细胞或植物细胞。在一些实施方案中,细胞来源于细胞系。在一些实施方案中,细胞是初生细胞。在一些实施方案中,细胞是免疫细胞。
在根据上述任一种方法的一些实施方案中,修饰包括靶核酸的位点特异性切割。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,修饰包括在靶基因座处引入选自插入、缺失和移码突变的突变。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,该方法还包括在允许供体dna在靶基因座处整合的条件下,使靶核酸与包含与靶基因座的序列同源的序列的供体dna接触。在一些实施方案中,供体dna编码选择标记,诸如报告蛋白。在一些实施方案中,该方法还包括评估细胞对选择标记的表达。在一些实施方案中,修饰包括在靶基因座处敲入外源序列,其中供体dna包含该外源序列。在一些实施方案中,修饰包括在靶基因座处引入置换突变,其中供体dna包含该置换突变。在一些实施方案中,置换突变是单核苷酸置换。在一些实施方案中,靶基因座是疾病相关的基因座。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,该方法包括在修饰之后对靶核酸测序。
在根据上述任一种方法的一些实施方案中,其中靶核酸存在于细胞中,修饰包括诱导细胞的表型变化。在一些实施方案中,该方法还包括评估细胞的表型变化。在一些实施方案中,修饰包括改变靶核酸的表达。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变。在一些实施方案中,靶基因座是疾病相关的基因座。
本申请的一个方面提供了一种组合物,该组合物包含含有ago蛋白和单链引导dna的复合物,其中复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1(诸如约1∶1)。
本申请的一个方面提供了一种递送系统,该递送系统包含含有ago蛋白和单链引导dna的复合物以及适于细胞内递送该复合物的载体,其中复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,载体选自细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物和纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1(诸如约1∶1)。
本申请的一个方面提供了一种试剂盒,该试剂盒包含编码ago蛋白的核酸和单链引导dna,其中ago蛋白和引导dna形成能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,编码ago蛋白的核酸对感兴趣的生物体是密码子优化的。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,引导dna在5′端被磷酸化。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,引导dna的长度为约10个至约50个核苷酸,诸如约15nt至约35nt、约20nt至约27nt、或约23nt至25nt。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个(诸如3个、2个、1个或0个的任一种)错配。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,靶基因座具有至少约60%(诸如至少约60%、65%、70%、75%、80%的任一种或更大)的gc含量。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。
在根据上述任一种组合物、递送系统或试剂盒的一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌(ferroglobusplacidus)、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白来源于解肝磷酯土地杆菌。在一些实施方案中,ago蛋白来源于微囊藻属。在一些实施方案中,ago蛋白来源于铜绿微囊藻。
在根据上述任一种组合物、递送系统或试剂盒的一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:2具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:11具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白包含与seqidno:41具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。
在根据上述任一种组合物、递送系统、或试剂盒的一些实施方案中,ago蛋白包括核定位信号(nls)。
在本申请的一个方面,提供了一种细胞,该细胞包含上述任一种组合物。
在本申请的一个方面,提供了一种试剂盒,该试剂盒包含上述任一种组合物或递送系统以及使用该试剂盒修饰靶核酸的说明书,其中靶核酸包含靶基因座。
根据随后的具体实施方式和所附权利要求书,本发明的这些方面和其他方面以及优点将变得显而易见。应当理解,本文所述的各个实施例的一种、一些或所有特性可组合,以形成本发明的其他实施例。
附图说明
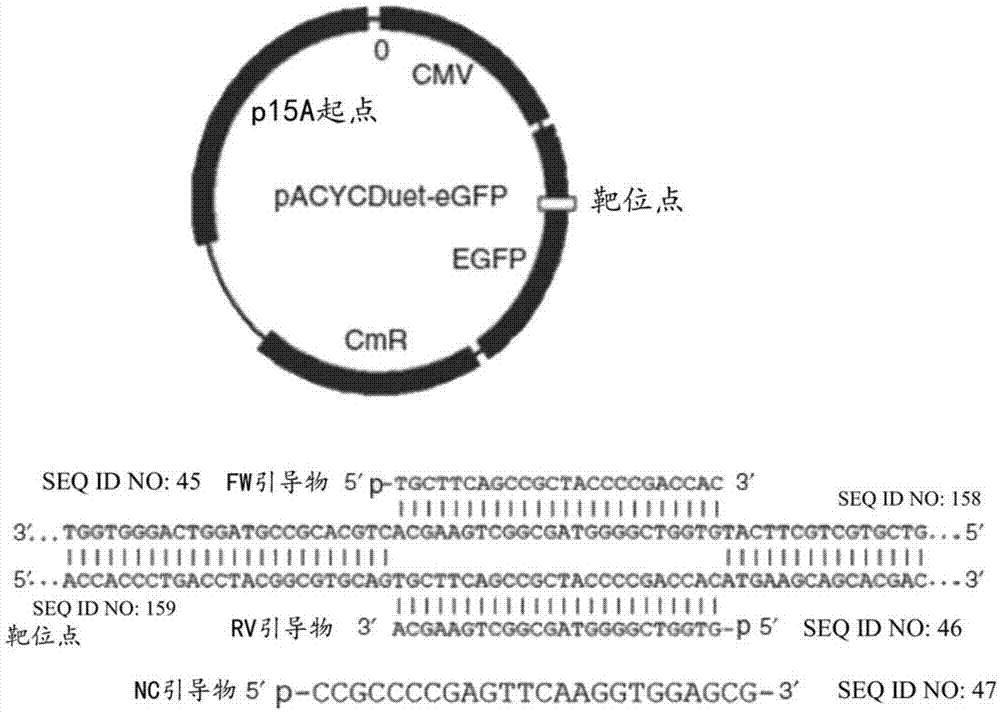
图1示出用于体外质粒切割测定的pacycduet-egfp质粒、靶位点和引导dna的示意性设计。fw和rv引导dna与质粒中的靶位点序列对准。还示出了5′-磷酸化nc(非特异性对照)引导dna的序列。
图2a示出靶pacycduet-egfp质粒在与从293t细胞纯化的ngago-fwgdna复合物一起温育4h、8h或72h后的电泳结果。
图2b示出靶pacycduet-egfp质粒与ngago-fwgdna复合物的混合物在不同温度下温育8小时的电泳结果。
图2c示出ngago-gdna复合物对质粒的切割产物的示例性序列和代表性测序色谱图。
图3a示出与或不与经纯化ngago-gdna复合物一起温育的线性化pacycduet-egfpdna的电泳结果。
图3b示出在37℃下86个核苷酸ssdna与或不与经纯化ngago-gdna复合物共温育8小时的电泳结果。
图4a示出靶pacycduet-egfp质粒与naago-fwgdna复合物的混合物在不同温度下温育8小时的电泳结果。
图4b示出靶pacycduet-egfp质粒与ntago-fwgdna复合物的混合物在不同温度下温育8小时的电泳结果。
图4c示出靶pacycduet-egfp质粒与maago-fwgdna复合物的混合物在不同温度下温育8小时的电泳结果。
图4d示出靶pacycduet-egfp质粒与syago-fwgdna复合物的混合物在不同温度下温育8小时的电泳结果。
图5示出ngago-6p-1质粒的示意图。
图6示出与大肠杆菌(e.coli)中所表达的gst-ngago共纯化的核酸的电泳结果。
图7a示出使用重新负载用于靶向pacycduet-egfp的各种ssdna引导物的ngago进行的体外质粒切割测定的电泳结果。
图7b示出在37℃或55℃下使用重新负载ssdna引导物的ngago在体外质粒切割测定1小时或72小时的电泳结果。在55℃下,引导物重新负载过程损害ngago的内切核酸酶活性。
图7c示出使用有或没有5′磷酸化的指定引导核酸进行体外质粒切割测定的电泳结果。
图8a示出结合ngago的核酸的电泳,其中ngago编码质粒被转染到293t细胞中,同时转染或不转染指定24-nt核酸。
图8b示出结合ngago的核酸的电泳,其中ngago编码质粒被转染到293t细胞中,随后转染5′磷酸化ssdna引导物(fw)。
图8c示出在293t细胞中表达,在表达48小时后提取,并与5′磷酸化ssdna引导物在55℃下一起温育1h或在37℃下温育8h的结合ngago的核酸的电泳。
图8d示出利用从293t细胞纯化的ngago进行体外质粒切割测定的电泳结果。在一些情况下,将293t细胞用ngago-编码质粒与靶互补引导物(fw)或随机引导物(nc)共转染。在一些情况下,将纯化的ngago或ngago-gdna复合物与靶互补引导物(fw)或随机引导物(nc)随后共温育。
图9示出ngago所遵循的“一种-引导物-忠实性”规则的示意图。当ngago表达质粒转染到哺乳动物细胞中以表达ngago蛋白时,ngago在与fw引导dna共温育时对切割无活性(顶部)。当ngago表达质粒与磷酸化或未磷酸化ssdna或ssrna引导物(fw引导物)共转染到哺乳动物细胞中时,仅5′磷酸化ssdna引导物被负载到ngago上以允许切割(中间)。当ngago表达质粒与非特异性引导物(nc引导物)共转染时,ngago-gdna复合物稍后不能负载第二特异性引导物(fw引导物)以激活切割活性(底部)。
图10示出ngago通过核定位信号(nls)定向至hela的细胞核。比例尺=100μm。图像代表20个独立的实验。
图11a示出靶质粒pegfp-n1的示意性设计,在指定的cmv启动子和egfp基因之内具有各种gdna的靶位点。
图11b示出靶质粒pegfp-n1上对于ngago的ssdna引导物和对于cas9的sgrna的杂交位点。
图11c示出用靶质粒pegfp-n1与ngago-表达质粒和指定ssdna引导物、或cas9-表达质粒和指定sgrna转录载体一起转染的hela细胞中egfp表达的western印迹杂交分析。结果代表三个独立的实验。基于egfp和肌动蛋白条带之间的密度测定比率的对应半定量结果显示于左下方的柱形图中。右侧柱形图示出由ngago-gdna系统与cas9-sgrna系统所引起的gfp表达水平倍数变化的统计分析(***表示p<0.01)。
图11d示出用靶质粒pegfp-n1、ngago编码质粒和各种长度的g3(n)引导物共转染的hela细胞中egfp表达的western印迹杂交分析。印迹代表三个独立的实验。
图12示出用靶质粒pegfp-n1、不同菌种的编码ago的各种质粒、以及g3gdna共转染的hela细胞中egfp表达的western印迹杂交分析。
图13a示出人dyrk1a基因的外显子11中的靶基因座和对应的引导dna的示意性设计。
图13b示出在dyrk1a基因座中显示ngago-gdna诱导的双链断裂的t7ei测定结果。
图13c示出在人dyrk1a基因中显示微缺失d10(缺失位点用省略号标记)的示例色谱图,以及使用g10引导物由克隆扩增子鉴定的突变等位基因的代表性序列。wt序列在顶部列出,并且g10引导物的dna靶序列以下划线标注。ngago-ssdna导致缺失(d10、d17、d25和d32)、突变(m1)、以及插入(+1、+2)。序列名称列于左侧,并根据序列变更类型(d,缺失;m,突变;+,插入),随之以变更的核苷酸数目进行命名。
图14a示出显示ngago/gdna引导物/靶和cas9/grna引导物/靶位置、t7ei切割位置和预测的pcr产物长度的示意性实验设计。
图14b示出显示所预测的ngago-gdna(g5和g10)和cas9-sgrna(sg-dyrk1a)的切割产物的t7ei测定结果。
图15a示出显示ngago-gdna在人基因组中指定的靶基因处诱导双链断裂的t7ei测定结果。
图15b示出显示ngago-gdna系统切割hba2、gata4、grin2b、hres1和apoe的三个独立t7ei实验的代表性印迹。
图15c示出ngago-gdna(g5-g18)在dyrk1a基因座中的切割效率,如由t7ei测定所测。
图15d示出ngago-gdna(g19-g26)在肌动蛋白基因座中的切割效率,如由t7ei测定所测。
图15e示出ngago-gdna(g27-g36)在emx1基因座中的切割效率,如由t7ei测定所测。
图16示出显示在各种哺乳动物细胞系中ngago-gdna诱导g10所靶向的dyrk1a基因座的双链断裂的t7ei测定结果。
图17a示出显示使用具有错配的经修饰24-nt长g10引导物,ngago-gdna诱导dyrk1a基因座的双链断裂的t7ei测定结果。
图17b示出显示使用具有错配的经修饰21-nt长g10引导物,ngago-gdna诱导dyrk1a基因座的双链断裂的t7ei测定结果。
图18a示出与对应的引导dna和sgrna对准的富gc人hba2基因座(顶部)和gata4基因座(底部)的序列。
图18b示出在人hba2和gata4基因的富gc基因座中比较ngago-gdna系统与cas9-sgrna系统之间的切割效率的t7ei测定结果。
图19a示出由ngago-gdna引发的hdr介导的供体dna插入的示意性设计。pcr引物区用箭头指示。
图19b示出了指示出hdr介导的供体dna成功插入靶定基因组基因座中的基因组pcr扩增子的序列色谱图。
图19c示出由ngago-gdna引发的nhej介导的突变的示意性设计。启动子(箭头)驱动mrfp表达,并跟随着靶位点和终止密码子。egfp在框外并且不表达(顶部)。在ngago-gdna-诱导的dsb和nhej-介导的移码突变之后,egfp在框内并且表达(表达)。
图19d示出用空载体、ngago-编码质粒、g52gdna、或ngago-编码质粒和g52gdna转染的mrfp-tga-egfp整合的细胞的流式细胞术分析。结果代表三个独立的实验。
图20a示出研究ngago-gdna或cas9-sgrna的脱靶基因组编辑效应的实验的示意性设计。
图20b示出显示cas9而非ngago的脱靶编辑的southern印迹杂交。
图21a示出ngago-gdna所引发的egfp供体dna插入dyrk1a基因座中的示意性实验设计。引物结合位置和g10位点用箭头指示。预期产物包括插入缺失、反向插入egfp供体dna(不表达gfp),以及egfp编码序列置于dyrk1a基因框内的正确插入构建体,从而允许gfp表达。正确的供体dna整合的基因座可包含g10位点的突变(g10′具有框以标注突变),这是ngago-gdna切割和g10位点的核苷酸移除的结果。
图21b示出靠近上游引物dy2的修饰dyrk1a基因座的pcr扩增子的测序色谱图。
图21c示出靠近介于dyrk1a基因座与插入的egfp之间的接头的修饰dyrk1a基因座的pcr扩增子的测序色谱图。顶部图示出修饰dyrk1a基因座中g10位点的突变。
图21d示出靠近下游引物g2r的修饰dyrk1a基因座的pcr扩增产物的测序色谱图。
图21e示出细胞在用ngago-gdna和egfp供体dna共转染之后的显微图像。左图示出相差显微图像。右图示出绿色通道荧光显微图像。
图22a示出ngago-gdna所介导的egfp供体dna敲入β-肌动蛋白基因座之后细胞的显微图像。左图示出绿色通道荧光显微图像,指示工程化蛋白质产物的表达。右图示出红色通道荧光显微图像,其中细胞中的f-肌动蛋白用tritc-鬼笔环肽染料染色。
图22b示出ngago-gdna所介导的egfp供体dna敲入β-肌动蛋白基因座之后细胞的显微图像。左图示出蓝色通道荧光显微图像,其中细胞核使用dapi染色。中间图示出绿色通道、红色通道和蓝色通道中细胞的显微图像的叠加。右图示出相差显微图像。
图22c示出靠近介于β-肌动蛋白基因座与插入的egfp之间的接头的修饰β-肌动蛋白基因座的pcr扩增子的测序色谱图。
图23a示出phago-gdna所介导的egfp供体dna敲入dyrk1a基因座之后细胞的显微图像。左图示出相差显微图像。右图示出绿色通道荧光显微图像。
图23b示出在phago-gdna所介导的egfp供体dna敲入β-肌动蛋白基因座之后细胞的显微图像。左上图示出绿色通道荧光显微图像,指示工程化蛋白质产物的表达。右上图示出红色通道荧光显微图像,其中细胞中的f-肌动蛋白用tritc-鬼笔环肽染料染色。下上图示出蓝色通道荧光显微图像,其中细胞核使用dapi染色。右下图示出相差显微图像。
图23c示出miago-gdna所介导的egfp供体dna敲入dyrk1a基因座之后细胞的显微图像。左图示出相差显微图像。右图示出绿色通道荧光显微图像。
图23d示出maago-gdna所介导的egfp供体dna敲入dyrk1a基因之后后细胞的显微图像。左图示出相差显微图像。右图示出绿色通道荧光显微图像。
图24a示出了5′磷酸盐结合位点的kqk基序周围的不同菌种的ago蛋白的序列比对。保守氨基酸残基kqk被突出显示。
图24b示出核酸酶活性位点的dde基序周围的不同菌种的ago蛋白的序列比对。保守氨基酸残基dde被突出显示。
图25示出来自不同菌种的ago蛋白的系统发育树。所示百分比是两个分支的共有序列之间的成对序列的同源性。用星号(*)标记的ago在与gdna一起转染到哺乳动物细胞中时不能切割靶核酸。
图26a示出在细菌宿主中表达的ngago-gdna对靶dna基因组的降解。
图26b示出ago-gdna在体外对线性化pgex-6p-1载体的降解。从用包含具有gst标签的ngago、miago、或maago表达盒的pgex-6p-1质粒转化的大肠杆菌细胞中纯化ago-gdna复合物。
具体实施方式
本发明提供了使用argonaute(ago)和引导dna(gdna)修饰靶核酸的方法、递送系统、组合物和试剂盒。本申请的一些实施方案提供了基于ago的基因或基因组编辑方法。本发明基于以下发现:某些ago蛋白(诸如,来自格氏嗜盐碱杆菌的ago,也称为“ngago”)可通过单链寡核苷酸dna(即gdna)引导而在生理温度下特异性识别和修饰具有gdna的互补序列的靶基因座。在一些实施方案中,ago-gdna诱导靶dna基因座的双链断裂(dsb),从而允许使用细胞dsb修复机制在dsb处进行基因编辑。在一些实施方案中,不诱导dna切割。本文所述ago-gdna方法的特征在于高特异性,并且可广泛应用于各种序列(包括具有高gc含量的序列)的靶基因座。本文所述方法的高特异性由ago-gdna系统的若干特征提供。在一些实施方案中,ago蛋白对gdna与靶基因座之间的错配具有较低耐受性(诸如最多3个错配)。在一些实施方案中,gdna与靶基因座之间的单个错配导致ago-gdna复合物的切割效率显著降低。在一些实施方案中,引导dna是5′磷酸化的。因为5′磷酸化短ssdna在哺乳动物细胞中较罕见,用此类ago-gdna系统来修饰哺乳动物细胞中的靶核酸使得由于细胞寡核苷酸错误引导ago蛋白而所致的非特异性修饰最小化。在一些实施方案中,引导dna仅可例如在ago蛋白表达期间负载到ago-gdna复合物中一次,并且一旦负载,ago蛋白就不能在生理温度(诸如37℃)下用另一游离ssdna替换其gdna。该特征在本文也被称为“一种-引导物-忠实性”规则,可进一步降低脱靶效应。此外,相比于用于其它基因编辑或沉默系统的引导rna,本发明方法的ssdna引导物可易于设计和制备,因为对于gdna,ago蛋白不具有任何特定序列或次级结构需求。将gdna转染到细胞中很简单,并且gdna的转染剂量可调节。可实现对靶核酸的多种修饰,包括但不限于位点特异性切割、插入缺失、敲除、敲入、置换(诸如单核苷酸置换)、以及基因表达改变。
因此,本申请的一个方面提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。
本申请的一个方面提供了一种组合物,该组合物包含含有ago蛋白和单链引导dna的复合物,其中复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。
本申请的一个方面提供了一种递送系统,该递送系统包含含有ago蛋白和单链引导dna的复合物以及适于细胞内递送该复合物的载体,其中复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。
本申请的一个方面提供了一种试剂盒,该试剂盒包含编码ago蛋白的核酸和单链引导dna,其中复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。
还提供了可用于本文所述方法的试剂盒和制品。
i.定义
除非另外指明,本发明涉及使用的科技术语将具有本领域的普通技术人员通常理解的含义。另外,除非上下文另有要求或明确指明,单数术语包括复数,复数术语也包括单数。
如本文所用,“argonaute”和“ago”可互换使用,并且指天然存在或工程化的蛋白质,其可经由单链寡核苷酸dna(即引导dna)引导来特异性识别包含引导dna的互补序列的靶核酸。某些ago蛋白在本文也被称为“argonaute核酸酶”,具有dna引导的内切核酸酶活性,即切割靶核酸中的内部磷酸二酯键。某些ago蛋白不切割靶核酸。
如本文所用,“引导dna”、“gdna”或“dna引导物”可互换地用于指能够与本申请的argonaute蛋白形成复合物并与靶核酸杂交的单链寡核苷酸dna。与引导dna杂交的靶核酸部分在本文中可互换地称为“靶基因座”或“靶位点”。结合引导dna的ago蛋白的复合物在本文称为“ago-gdna”或“ago-g”。
如本文所用,“供体dna”是指可整合进ago-gdna复合物所诱导的双链断裂的位点中的多核苷酸。
术语“核酸”、“多核苷酸”和“核苷酸序列”可互换地用于指任何长度核苷酸的聚合物形式,包括脱氧核糖核苷酸、核糖核苷酸、它们的组合、以及它们的类似物。“寡核苷酸”和“寡”可互换地用于指具有不超过约50个核苷酸的短多核苷酸。
“互补性”是指核酸通过传统的沃森-克里克或其它非传统类型与另一种核酸形成氢键的能力。互补百分比表示核酸分子中可与第二核酸形成氢键(例如沃森-克里克碱基配对)的残基的百分比(例如,10个中有约5个、6个、7个、8个、9个、10个,分别为约50%、60%、70%、80%、90%和100%互补性)。“完全互补”意指核酸序列中的所有连续残基与第二核酸序列中相同数目的连续残基氢键结合。如本文所用,“基本上互补”是指在约8个、9个、10个、11个、12个、13个、14个、15个、16个、17个、18个、19个、20个、21个、22个、23个、24个、25个、30个、35个、40个、45个、50个、或更多个核苷酸的区域上至少约60%、65%、70%、75%、80%、85%、90%、95%、97%、98%、99%、或100%的任一种的互补程度,或者指在严格条件下杂交的两个核酸。
“错配”是指第一核酸中不与第二核酸中对应的核苷酸形成传统的沃森克里克碱基对的核苷酸。
如本文所用,“靶定”或“靶向”是指gdna或ago-gdna复合物特异性结合核酸。“特异性结合”是指在严格条件下杂交。
如本文所用,用于杂交的“严格条件”是指与靶序列有互补性的核酸主要与靶序列杂交,并且基本上不与非靶序列杂交的条件。严格条件通常是序列依赖性的,并根据多个因素变化。一般来讲,序列越长,序列特异性杂交其靶序列的温度就越高。严格条件的非限制性示例详细描述于tijssen(1993),laboratorytechniquesinbiochemistryandmolecularbiology-hybridizationwithnucleicacidprobesparti,第二章“overviewofprinciplesofhybridizationandthestrategyofnucleicacidprobeassay”,elsevier,n,y。
“杂交”是指一个或多个多核苷酸反应形成经由核苷酸残基的碱基之间的氢键合而稳定化的复合物的反应。氢键可以通过沃森-克里克碱基配对、hoogstein结合,或以任何其它序列特异性方式产生。复合物可包含形成双链体结构的两条链、形成多链复合物的三条或更多条链、自杂交单链、或这些链的任何组合。杂交反应可构成更广泛过程中的一步,诸如pcr的引发,或酶对多核苷酸的切割。能够与给定序列杂交的序列称为给定序列的“互补序列”。
相对于肽、多肽或蛋白质序列,“序列同一性百分比(%)”定义为在比对序列和引入缺口(如果需要)以实现最大序列同一性百分比之后,候选序列中与特异性肽或多肽序列中氨基酸残基相同的氨基酸残基的百分比。相对于肽、多肽或蛋白质序列,“序列同源性百分比(%)”是在比对序列和引入缺口(如果需要)以实现最大序列同源性百分比之后,候选序列中相对于特异性肽或多肽序列中氨基酸残基相同或保守置换的氨基酸残基的百分比。为确定氨基酸序列同一性百分比所进行的比对可用本领域技术范围内的各种方式获得,例如使用公众可获取的计算机软件,如blast、blast-2、align或megaligntm(dnastar)软件。本领域的技术人员可确定用于测量比对的适当参数,包括在被比较序列的全长上实现最大比对所需的任何算法。
术语“多肽”和“肽”在本文中可互换地用于指任何长度氨基酸的聚合物。该聚合物可为直链或支链,其可包含修饰氨基酸,并且其可间杂有非氨基酸。蛋白质可具有一种或多种多肽。该术语还涵盖经过修饰的氨基酸聚合物;例如,二硫键形成、糖基化、脂化、乙酰化、磷酸化、或任何其它操纵,诸如与标记组分缀合。
术语“转染”、“转化”和“递送”在本文中可互换地用于指外源分子(诸如核酸、蛋白质或它们的复合物)转移或引入细胞内的过程。
术语“细胞”包括原代目标细胞及其子代。术语“宿主细胞”是指其中已引入外源核酸或蛋白质复合物(诸如ago-gdna复合物)的细胞,包括此类细胞的子代。细胞和宿主细胞包括“转化体”和“转化细胞”,其包括原代转化细胞和由其衍生的子代,而不考虑传代数。子代的核酸内容物可能不与亲本细胞完全相同,但可包含突变。与原始转化细胞中所筛选或选择的功能或生物活性相同的突变子代包括在本文之内。
如本文所用,术语“分离的”可指经由人工脱离其天然环境而存在,因此不是天然产物的核酸或多肽。分离的核酸或多肽可以纯化形式存在,并且/或者可存在于非天然环境中,例如转基因细胞中。
应当理解,本文所述的本发明的实施方案包括“由...组成”和/或“基本由...组成”实施方案。
本文提及“约”的值或参数包括(并描述)涉及该值或参数本身的变化。例如,关于“约x”的描述包括对“x”的描述。
如本文所使用,提及“非”某个值或参数通常表示并描述“除该值或参数之外”的值或参数。
本文所用的术语“约x-y”具有与“约x至约y”相同的含义。
如本文所用,单数形式“一”、“或”、和“该”包括复数指代,除非上下文另外明确规定。
除非相反地特别说明,否则本发明的实践将采用本领域技术范围内的病毒学、免疫学、微生物学、分子生物学和重组dna技术的常规方法,其中许多出于说明性目的在下文进行描述。此类技术在文献中作出了充分地说明。参见例如currentprotocolsinmolecularbiologyorcurrentprotocolsinimmunology,johnwiley&sons,newyork,n.y.(2009);ausubel等人,shortprotocolsinmolecularbiology,第3版,wiley&sons,1995;sambrook和russell,molecularcloning:alaboratorymanual(第3版,2001);maniatis等人molecularcloning:alaboratorymanual(1982);dnacloning:apracticalapproach,vol.i&ii(d.glover编辑);oligonucleotidesynthesis(n.gait编辑,1984);nucleicacidhybridization(b.hames&s.higgins编辑,1985);transcriptionandtranslation(b.hames&s.higgins编辑,1984);animalcellculture(r.freshney编辑,1986);perbal,apracticalguidetomolecularcloning(1984)以及其它类似的文献。
ii.修饰把核酸的方法
本申请提供了使用argonaute和单链引导dna修饰靶核酸的方法。该方法可用于体外或细胞内基因编辑、位点特异性切割、基因沉默、以及其它核酸修饰。
本申请的一个方面提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna在5′端被磷酸化。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和单链引导dna接触,其中ago蛋白和引导dna形成特异性识别并切割靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna在5′端被磷酸化。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和单链引导dna接触,其中ago蛋白和所述引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,并且其中靶基因座是包含与引导dna的序列互补的序列的双链dna。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna在5′端被磷酸化。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别并切割靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,并且其中靶基因座是包含与引导dna的序列互补的序列的双链dna。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列,并且其中ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列,并且其中ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是包含与引导dna的序列互补的序列的双链dna,并且其中ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是包含与引导dna的序列互补的序列的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列,并且其中ago蛋白来源于铜绿微囊藻。在一些实施方案中,ago蛋白包含与seqidno:2具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是包含与引导dna的序列互补的序列的双链dna,并且其中ago蛋白来源于铜绿微囊藻。在一些实施方案中,ago蛋白包含与seqidno:2具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列,并且其中ago蛋白来源于微囊藻属。在一些实施方案中,ago蛋白包含与seqidno:11具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是包含与引导dna的序列互补的序列的双链dna,并且其中ago蛋白来源于微囊藻属。在一些实施方案中,ago蛋白包含与seqidno:11具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,其中靶基因座包含与引导dna的序列互补的序列,并且其中ago蛋白来源于解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与seqidno:41具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是包含与引导dna的序列互补的序列的双链dna,并且其中ago蛋白来源于解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与seqidno:41具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
本文所述的方法可用于在体外和细胞内修饰靶核酸。在一些实施方案中,靶核酸是分离的dna。在一些实施方案中,靶核酸是质粒。在一些实施方案中,靶核酸是包含双链dna区域的分离核酸。本文所述的argonaute核酸酶可用于在体外将位点特异性切割引入靶核酸(诸如双链dna)中。切割位点处于引导dna杂交到其上的靶核酸区域内(即“靶基因座”)。值得注意的是,本文所述的argonaute核酸酶可使用单一引导dna来诱导双链dna(诸如质粒)的双链断裂。序列与靶位点的序列互补(诸如完全互补或基本上互补)的引导dna可用于在靶核酸中的靶位点处诱导双链断裂。
限制性酶通常用于在体外位点特异性切割核酸。不同于限制性酶,argonaute核酸酶不需要切割位点以包含回文序列。因为本文所述的argonaute核酸酶不具有序列偏好,通过设计具有合适序列的引导dna,本文所述的方法可用于切割靶核酸中的任何位点,包括富gc位点。另外,因为引导dna的长度长于识别序列,用于位点特异性切割的基于ago-gdna的方法比限制性酶的特异性更高且脱靶效应更低。值得注意的是,本文所述的argonaute核酸酶不具有外切核酸酶活性。
因此,在一些实施方案中,提供了一种在体外位点特异性切割靶核酸(诸如质粒)的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和单链引导dna(诸如5′磷酸化单链引导dna)接触,其中ago蛋白和引导dna形成特异性识别并切割靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,提供了一种在体外位点特异性切割靶核酸(诸如质粒)的方法,包括在约10℃至约60℃(诸如约37℃)的温度下使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是包含与引导dna的序列互补的序列的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,接触在二价金属离子(诸如至少约0.1mm)的存在下进行。
在一些实施方案中,靶核酸存在于细胞中。靶核酸可为细胞中的内源核酸(诸如基因组dna或rna)、或细胞中的外源核酸,诸如病毒核酸。本文所述的方法与多种细胞兼容,包括原核细胞和真核细胞两者。因为非嗜热菌种的生理温度在约10℃至约60℃的范围内,先前已知具有dna引导的核酸内切酶活性的argonaute核酸酶,诸如嗜热栖热菌的argonaute,不能在非嗜热菌种的活细胞内切割靶核酸。参见例如wo2014/189628。此处,本申请的发明人鉴定出可由单链引导dna引导而在生理温度(诸如约10℃至约60℃,或约37℃)下特异性识别和/或切割细胞内靶核酸的argonaute,从而实现本文所述的修饰细胞内靶核酸的方法。
因此,在一些实施方案中,提供了一种修饰细胞中靶核酸的方法,包括使靶核酸与argonaute(ago)蛋白和单链引导dna(诸如5′磷酸化单链引导dna)接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,靶核酸对于细胞是内源性的。在一些实施方案中,靶核酸是基因组dna。在一些实施方案中,靶核酸是rna,诸如mrna。在一些实施方案中,靶核酸对于细胞是外源性的。在一些实施方案中,靶核酸被整合进细胞的基因组中。在一些实施方案中,靶核酸未整合进细胞的基因组中。在一些实施方案中,靶核酸是病毒dna。在一些实施方案中,细胞是原核细胞。在一些实施方案中,细胞是真核细胞,诸如植物、真菌、酵母或哺乳动物细胞。在一些实施方案中,细胞是人细胞。在一些实施方案中,细胞来源于细胞系。在一些实施方案中,细胞是初生细胞。在一些实施方案中,细胞是免疫细胞。
在一些实施方案中,提供了一种修饰细胞中靶核酸的方法,包括使靶核酸与argonaute(ago)蛋白和5′磷酸化单链引导dna接触,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白和引导dna存在于预形成的复合物中。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,靶核酸对于细胞是内源性的。在一些实施方案中,靶核酸是基因组dna。在一些实施方案中,靶核酸是rna,诸如mrna。在一些实施方案中,靶核酸对于细胞是外源性的。在一些实施方案中,靶核酸被整合进细胞的基因组中。在一些实施方案中,靶核酸未整合进细胞的基因组中。在一些实施方案中,靶核酸是病毒dna。在一些实施方案中,细胞是原核细胞。在一些实施方案中,细胞是真核细胞,诸如植物、真菌、酵母或哺乳动物细胞。在一些实施方案中,细胞是人细胞。在一些实施方案中,细胞来源于细胞系。在一些实施方案中,细胞是初生细胞。在一些实施方案中,细胞是免疫细胞。
可通过本领域已知的任何方法将ago蛋白和引导dna递送到细胞中。因为本文所述的一些ago蛋白遵循“一种-引导物-忠实性”规则,即一旦ago蛋白与引导dna形成复合物,则在低于约50℃(诸如约37℃)的温度下ago蛋白就不与引导dna解离或者用未结合的引导dna交换该引导dna,从而确保方法的功效,ago蛋白和引导dna可作为预形成的复合物递送到细胞中,或者ago蛋白在引导dna存在下由细胞表达。在后一种情况下,可将编码ago蛋白的核酸转染到细胞中以允许ago蛋白被细胞表达。引导dna可在编码ago蛋白的核酸之前或与其同时转染到细胞中。在一些实施方案中,在将ago(蛋白质或核酸)和引导dna引入细胞之后,用gdna进一步转染细胞一次或多次(诸如约1、2、3、4次的任一种、或更多次)。
因此,在一些实施方案中,提供了一种修饰细胞中靶核酸的方法,包括将单链引导dna(诸如5′磷酸化单链引导dna)和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。
在一些实施方案中,提供了一种修饰细胞中靶核酸的方法,包括使5′磷酸化单链引导dna和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。
在一些实施方案中,提供了一种修饰细胞中靶核酸的方法,包括将包含ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。
在一些实施方案中,提供了一种修饰细胞中靶核酸的方法,包括将包含ago蛋白和5′磷酸化单链引导dna的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb),其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。
本文所述的ago蛋白可以多种方式修饰细胞中的靶核酸。在一些实施方案中,该方法在靶核酸中诱导位点特异性切割。在一些实施方案中,该方法切割细菌细胞中的基因组dna。在一些实施方案中,该方法切割细胞中的病毒核酸。在一些实施方案中,该方法改变(诸如提高或降低)细胞中靶核酸的表达水平。在一些实施方案中,该方法使细胞中靶核酸的表达水平降低或沉默。在一些实施方案中,该方法在细胞中使用一种或多种内源性dna修复途径,诸如非同源末端连接(nhej)和同源定向重组(hdr),以利用ago蛋白修复靶基因座中诱导的双链断裂,从而在靶基因座处引入突变或外源序列。在一些实施方案中,该方法在靶基因座处引入突变。示例性突变包括但不限于插入、缺失、置换和移码。在一些实施方案中,该方法在靶位点插入供体dna。在一些实施方案中,供体dna的插入导致选择标记或报告蛋白引入细胞中。在一些实施方案中,供体dna的插入导致基因敲入。在一些实施方案中,供体dna的插入导致基因敲除突变。在一些实施方案中,供体dna的插入导致置换突变,诸如单核苷酸置换。在一些实施方案中,该方法诱导细胞的表型变化。
因此,在一些实施方案中,提供了一种在细胞内位点特异性切割靶核酸(诸如病毒核酸)的方法,包括将单链引导dna(诸如5′磷酸化单链引导dna)和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性切割靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内位点特异性切割靶核酸(诸如病毒核酸)的方法,包括使5′磷酸化单链引导dna和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内位点特异性切割靶核酸(诸如病毒核酸)的方法,包括将包含ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的预形成的复合物递送到细胞中,其中复合物特异性切割靶核酸中的靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内位点特异性切割靶核酸(诸如病毒核酸)的方法,包括将包含ago蛋白和5′磷酸化单链引导dna的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb),其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种抑制靶细胞(诸如细菌细胞)生长的方法,包括使以下项转染到靶细胞中:(a)编码ago蛋白的核酸,以及(b)靶向靶细胞的基因组dna的包含一个或多个引导dna的载体,其中ago蛋白和引导dna形成特异性识别并切割基因组dna中的一个或多个靶基因座的复合物,并且其中靶基因座包含一个或多个引导dna的互补序列。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,使载体和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,载体在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,载体在转染之前是线性化的。在一些实施方案中,编码ago蛋白的核酸对靶细胞是密码子优化的。
在一些实施方案中,提供了一种使细胞中靶核酸(诸如基因)的表达改变(诸如降低)的方法,包括使单链引导dna(诸如5′磷酸化单链引导dna)和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种使细胞中靶核酸(诸如基因)的表达改变(诸如降低)的方法,包括使5′磷酸化单链引导dna和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种使细胞中靶核酸(诸如基因)的表达改变(诸如降低)的方法,包括将包含ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种使细胞中靶核酸(诸如基因)的表达改变(诸如降低)的方法,包括将包含ago蛋白和5′磷酸化单链引导dna的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb),其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中引入突变(诸如插入缺失或移码突变)的方法,包括将单链引导dna(诸如5′磷酸化单链引导dna)和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双键断裂的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中引入突变(诸如插入缺失或移码突变)的方法,包括使5′磷酸化单链引导dna和编码ago蛋白的核酸转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中引入突变(诸如插入缺失或移码突变)的方法,包括将包含ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂,并且其中靶基因座是所包含的序列与引导dna的序列互补的双链dna。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中引入突变(诸如插入缺失或移码突变)的方法,包括将包含ago蛋白和5′磷酸化单链引导dna的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb),其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。
在一些实施方案中,该方法还包括在允许供体dna在靶基因座处整合的条件下,使靶核酸与包含与靶基因座的序列的同源的序列的供体dna接触。可使用本领域已知的任何方法将供体dna递送到细胞中。在一些实施方案中,将供体dna与编码ago蛋白的核酸和/或引导dna同时地、或与编码ago蛋白的核酸和/或引导dna依序地(例如在其之后)递送到细胞中。在一些实施方案中,将供体dna与包含ago蛋白和引导dna的预形成的复合物同时地,或与预形成的复合物依序地(例如之前或之后)递送到细胞中。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中插入供体dna的方法,包括将单链引导dna(诸如5′磷酸化单链引导dna)、编码ago蛋白的核酸、以及供体dna转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双键断裂的复合物,其中供体dna整合在靶基因座中的dsb处,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中供体dna包含与靶基因座的序列同源的序列。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。在一些实施方案中,其中供体dna包含外源序列(诸如外源基因),该方法在靶基因座处引入外源序列的敲入。在一些实施方案中,其中供体dna包含置换突变(诸如单核苷酸置换),该方法在靶基因座处引入置换突变。在一些实施方案中,该方法在靶基因座处引入基因敲除突变。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中插入供体dna的方法,包括将5′磷酸化单链引导dna、编码ago蛋白的核酸、以及供体dna转染到细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双键断裂(dsb)的复合物,其中供体dna整合在靶基因座中的dsb处,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,其中供体dna包含与靶基因座的序列同源的序列,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到细胞中。在一些实施方案中,用引导dna转染细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对细胞所来源的生物体是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。在一些实施方案中,其中供体dna包含外源序列(诸如外源基因),该方法在靶基因座处引入外源序列的敲入。在一些实施方案中,其中供体dna包含置换突变(诸如单核苷酸置换),该方法在靶基因座处引入置换突变。在一些实施方案中,该方法在靶基因座处引入基因敲除突变。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中插入供体dna的方法,包括将供体dna以及包含ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双键断裂,其中供体dna整合在靶基因座中的dsb处,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中供体dna包含与靶基因座的序列同源的序列。在一些实施方案中,其中在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。在一些实施方案中,其中供体dna包含外源序列(诸如外源基因),该方法在靶基因座处引入外源序列的敲入。在一些实施方案中,其中供体dna包含置换突变(诸如单核苷酸置换),该方法在靶基因座处引入置换突变。在一些实施方案中,该方法在靶基因座处引入基因敲除突变。
在一些实施方案中,提供了一种在细胞内的靶核酸(诸如基因组dna)中插入供体dna的方法,包括将供体dna以及包含ago蛋白和5′磷酸化单链引导dna的预形成的复合物递送到细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双键断裂(dsb),其中供体dna整合在靶基因座中的dsb处,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,其中供体dna包含与靶基因座的序列同源的序列,并且其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理细胞。在一些实施方案中,细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,该方法诱导细胞的表型变化。在一些实施方案中,其中供体dna包含外源序列(诸如外源基因),该方法在靶基因座处引入外源序列的敲入。在一些实施方案中,其中供体dna包含置换突变(诸如单核苷酸置换),该方法在靶基因座处引入置换突变。在一些实施方案中,该方法在靶基因座处引入基因敲除突变。
在一些实施方案中,该方法包括在修饰后对细胞进行评估的一个或多个步骤。在一些实施方案中,该方法包括评估细胞的表型变化。在一些实施方案中,其中供体dna包含选择标记诸如报告蛋白,该方法包括评估细胞对选择标记的表达。在一些实施方案中,该方法包括对靶核酸测序。在一些实施方案中,该方法包括修饰多个细胞中的靶核酸,并基于以下项的一者或多者选择具有修饰的靶核酸的细胞:(1)细胞的表型变化;(2)选择标记诸如报告蛋白的表达,其中供体dna包含选择标记;和/或(3)细胞中修饰靶核酸的序列。
因此,在一些实施方案中,提供了一种修饰多个细胞中的靶核酸的方法,包括:(a)将单链引导dna(诸如5′磷酸化单链引导dna)和编码ago蛋白的核酸转染到多个细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列;并且(b)基于以下项的一者或多者选择具有修饰的靶核酸的细胞:(1)细胞的表型变化;(2)选择标记(诸如报告蛋白)的表达,其中该方法还包括在允许供体dna在靶基因座处整合的条件下,使靶核酸与包含与靶基因座的序列同源的序列的供体dna接触,并且其中供体dna包含选择标记;和/或(3)细胞中修饰靶核酸的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,多个细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到多个细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到多个细胞中。在一些实施方案中,用引导dna转染多个细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对多个细胞所来源的菌种是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理多个细胞。在一些实施方案中,多个细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,修饰包括在靶基因座引入突变(诸如插入缺失或移码突变)。在一些实施方案中,修饰包括改变靶核酸的表达。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变。在一些实施方案中,修饰包括诱导细胞的表型变化。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变、外源序列敲入、或置换(诸如单核苷酸置换)突变。
在一些实施方案中,提供了一种包括修饰多个细胞中靶核酸的方法,包括:(a)使5′磷酸化单链引导dna和编码ago蛋白的核酸转染到多个细胞中,其中ago蛋白和引导dna形成特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb)的复合物,其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌;并且(b)基于以下项的一者或多者选择具有修饰的靶核酸的细胞:(1)细胞的表型变化;(2)选择标记(诸如报告蛋白)的表达,其中该方法还包括在允许供体dna在靶基因座处整合的条件下,使靶核酸与包含与靶基因座的序列同源的序列的供体dna接触,并且其中供体dna包含选择标记;和/或(3)细胞中修饰靶核酸的序列。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,多个细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,使引导dna和编码ago蛋白的核酸同时转染到多个细胞中。在一些实施方案中,引导dna在编码ago蛋白的核酸之前转染到多个细胞中。在一些实施方案中,用引导dna转染多个细胞至少两次(诸如约2次、3次、4次、5次、6次的任一种或更多次)。在一些实施方案中,引导dna与编码ago蛋白的核酸的摩尔比为至少约100∶1。在一些实施方案中,编码ago蛋白的核酸对多个细胞所来源的菌种是密码子优化的。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理多个细胞。在一些实施方案中,多个细胞未被(其它)非病毒微生物污染。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒(诸如至少约95%超螺旋)。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,修饰包括在靶基因座引入突变(诸如插入缺失或移码突变)。在一些实施方案中,修饰包括改变靶核酸的表达。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变。在一些实施方案中,修饰包括诱导细胞的表型变化。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变、外源序列敲入、或置换(诸如单核苷酸置换)突变。
在一些实施方案中,提供了一种修饰多个细胞中靶核酸的方法,包括:(a)将包含ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的预形成的复合物递送到多个细胞中,其中复合物特异性识别靶核酸中的靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列;并且(b)基于以下项的一者或多者选择具有修饰的靶核酸的细胞:(1)细胞的表型变化;(2)选择标记(诸如报告蛋白)的表达,其中该方法还包括在允许供体dna在靶基因座处整合的条件下,使靶核酸与包含与靶基因座的序列同源的序列的供体dna接触,并且其中供体dna包含选择标记;和/或(3)细胞中修饰靶核酸的序列。在一些实施方案中,复合物切割靶基因座。在一些实施方案中,复合物不切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白诱导靶基因座的双链断裂。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,多个细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理多个细胞。在一些实施方案中,多个细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到多个细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到多个细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,修饰包括在靶基因座引入突变(诸如插入缺失或移码突变)。在一些实施方案中,修饰包括改变靶核酸的表达。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变。在一些实施方案中,修饰包括诱导细胞的表型变化。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变、外源序列敲入、或置换(诸如单核苷酸置换)突变。
在一些实施方案中,提供了一种修饰多个细胞中靶核酸的方法,包括:(a)将包含ago蛋白和5′磷酸化单链引导dna的预形成的复合物递送到多个细胞中,其中复合物特异性识别靶核酸中的靶基因座并诱导靶基因座的双链断裂(dsb),其中靶基因座是所包含的序列与引导dna的序列互补的双链dna,并且其中ago蛋白来源于格氏嗜盐碱杆菌;并且(b)基于以下项的一者或多者选择具有修饰的靶核酸的细胞:(1)细胞的表型变化;(2)选择标记(诸如报告蛋白)的表达,其中该方法还包括在允许供体dna在靶基因座处整合的条件下,使靶核酸与包含与靶基因座的序列同源的序列的供体dna接触,并且其中供体dna包含选择标记;和/或(3)细胞中修饰靶核酸的序列。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%序列同源性(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)的氨基酸序列。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna的长度为约10个至约50个(诸如约20个至约30个)核苷酸。在一些实施方案中,多个细胞补充有二价金属离子(诸如至少约0.1mm)。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,用一种或多种抗生素处理多个细胞。在一些实施方案中,多个细胞未被(其它)非病毒微生物污染。在一些实施方案中,复合物中引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到多个细胞中。在一些实施方案中,预形成的复合物经由选自以下的载体递送到多个细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,修饰包括在靶基因座引入突变(诸如插入缺失或移码突变)。在一些实施方案中,修饰包括改变靶核酸的表达。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变。在一些实施方案中,修饰包括诱导细胞的表型变化。在一些实施方案中,修饰包括在靶基因座处引入基因敲除突变、外源序列敲入、或置换(诸如单核苷酸置换)突变。
在一些实施方案中,提供了argonaute核酸酶在基因编辑中的用途,其中argonaute核酸酶能够使用5′磷酸化单链寡核苷酸dna作为引导物在约10℃-60℃下切割双链靶dna并诱导靶dna的双链断裂(dsb)。在一些实施方案中,提供了argonaute核酸酶在基因编辑中的用途,其中argonaute核酸酶利用5′磷酸化单链寡核苷酸dna作为引导物并与引导dna形成复合物,其中复合物切割包含引导dna的互补序列的双链靶dna的两条链,从而提供靶dna的双链断裂。在一些实施方案中,其中靶dna是细胞内靶基因,该用途包括在细胞中使用内源性非同源末端连接(nhej)修复途径来修复靶基因的双链断裂,从而引入突变以敲除或修饰靶基因或其功能区。另选地,在一些实施方案中,其中靶dna是细胞内靶基因,该用途包括在包含靶基因的同源序列的外源dna片段存在下,在细胞中使用内源性同源定向重组(hdr)途径来修复靶基因的双链断裂,从而将外源dna片段插入dsb中以实现靶基因或其功能区的专有修饰。
在上述任一种用途的一些实施方案中,argonaute核酸酶包含图24a-24b中6个关键保守氨基酸残基中的至少5个,其中6个关键保守残基包括mid结构域的5′磷酸盐结合位点中的3个保守残基,以及piwi结构域的核酸酶活性位点中的3个保守残基。在一些实施方案中,argonaute核酸酶是选自以下项的argonaute核酸酶:(1)具有ncbi登录号afz73749.1的argonaute核酸酶(seqidno:1);(2)具有genbank登录号elz29017.1的argonaute核酸酶(seqidno:3);(3)具有ncbi登录号wp_006111085.1的argonaute核酸酶(seqidno:4);(4)具有ncbi登录号wp_006090832.1的argonaute核酸酶(seqidno:5);(5)具有ncbi登录号wp_012265209.1的argonaute核酸酶(seqidno:2);(6)具有ncbi登录号wp_006183335.1的argonaute核酸酶(seqidno:6);(7)具有ncbi登录号wp_006054116.1的argonaute核酸酶(seqidno:7);(8)具有ncbi登录号wp_048159825.1的argonaute核酸酶(seqidno:8);(9)具有ncbi登录号wp_011056792.1的argonaute核酸酶(seqidno:9);(10)具有ncbi登录号wp_012659190.1的argonaute核酸酶(seqidno:10);(11)具有ncbi登录号wp_002747795.1的argonaute核酸酶(seqidno:11);(12)具有ncbi登录号wp_011378069.1的argonaute核酸酶(seqidno:12);(13)具有genbank登录号cda11056.1的argonaute核酸酶(seqidno:13);(14)具有ncbi登录号wp_003477422.1的argonaute核酸酶(seqidno:14);(15)具有ncbi登录号wp_016205751.1的argonaute核酸酶(seqidno:15);(16)具有genbank登录号cdb74854.1的argonaute核酸酶(seqidno:16);(17)具有ncbi登录号wp_007287731.1的argonaute核酸酶(seqidno:17);(18)具有ncbi登录号wp_012966655.1的argonaute核酸酶(seqidno:18);(19)具有genbank登录号ahg02841.1的argonaute核酸酶(seqidno:19);(20)具有ncbi登录号wp_015791216.1的argonaute核酸酶(seqidno:20);(21)具有ncbi登录号wp_019364073.1的argonaute核酸酶(seqidno:21);(22)具有genbank登录号afk51052.1的argonaute核酸酶(seqidno:22);(23)具有ncbi登录号wp_011238781.1的argonaute核酸酶(seqidno:23);(24)具有ncbi登录号wp_012306644.1的argonaute核酸酶(seqidno:24);(25)具有ncbi登录号wp_012572468.1的argonaute核酸酶(seqidno:25);(26)具有genbank登录号aam02524.1的argonaute核酸酶(seqidno:26);(27)具有ncbi登录号wp_011244830.1的argonaute核酸酶(seqidno:27);(28)具有genbank登录号abd00306.1的argonaute核酸酶(seqidno:28);(29)具有ncbi登录号wp_012575214.1的argonaute核酸酶(seqidno:29);(30)具有genbank登录号acq71053.1的argonaute核酸酶(seqidno:30);(31)具有genbank登录号bad80710.1的argonaute核酸酶(seqidno:31);(32)具有genbank登录号abb57564.1的argonaute核酸酶(seqidno:32);(33)具有genbank登录号eaw33836.1的argonaute核酸酶(seqidno:33);(34)具有genbank登录号abd03669.1的argonaute核酸酶(seqidno:34);(35)具有genbank登录号als17562.1的argonaute核酸酶(seqidno:35);以及(36)具有ncbi登录号wp_049912037.1的argonaute核酸酶(seqidno:36)。
在上述任一种用途中的一些实施方案中,argonaute核酸酶与以上公开的36种argonaute核酸酶中的任一种有至少约80%的序列同源性,并且argonaute核酸酶能够使用5′磷酸化单链寡核苷酸dna作为引导物在约10℃-60℃(优选约37℃)下切割双链靶dna的两条链并诱导双链断裂。在一些优选的实施方案中,argonaute核酸酶与以上公开的36种argonaute核酸酶中的任一种有至少约90%的序列同源性,诸如至少95%的序列同源性。
在一些实施方案中,提供了编码36种argonaute核酸酶中任一种或与其具有至少约80%的序列同源性的argonaute核酸酶的基因序列、包含该基因序列的质粒或病毒载体、或包含argonaute核酸酶和5′磷酸化单链寡核苷酸dna的蛋白质复合物在基因编辑(包括靶向编辑细胞内病毒dna、体外靶向编辑dna、以及靶向编辑染色体dna)中的用途。还提供了编码argonaute核酸酶的基因序列在基因编辑中的用途,表达载体的构建、或基因编辑试剂盒的制备。
本申请提供了argonaute核酸酶、编辑argonaute核酸酶的基因序列、其质粒、或蛋白质复合物在靶向基因组编辑中的用途,其中argonaute核酸酶诱导基因组的双链断裂时,并在具有同源序列的外源dna片段存在下,外源dna片段可经由细胞内源性同源定向重组途径插入双链断裂中,从而实现靶基因或其功能区的专有修饰。
a.argonaute
本文所述的方法所基于的argonaute能够使用单链引导dna在约10℃至约60℃(诸如约10℃至约20℃、约20℃至约30℃、约30℃至约40℃、约40℃至约50℃、约50℃至约60℃、约20℃至约40℃、约10℃至约50℃、或约15℃至约45℃的任一种)的温度下特异性识别包含引导dna的完全或基本互补序列的靶核酸。在一些实施方案中,argonaute在约10℃、15℃、20℃、25℃、30℃、32℃、34℃、36℃、37℃、38℃、或40℃的任一种下呈活性。在一些实施方案中,argonaute的活性在至少约50℃下降低至少约10%、20%、30%、40%、50%、60%、70%、80%、90%的任一种或更多。
argonaute的活性包括靶基因座的特异性结合和切割。在一些实施方案中,argonaute是切割与引导dna杂交的靶基因座处的靶核酸的内切核酸酶。在一些实施方案中,ago-gdna复合物切割双链靶dna的一条链。在一些实施方案中,ago-gdna复合物切割双链靶dna的两条链。在一些实施方案中,ago-gdna复合物切割rna,诸如mrna。在一些实施方案中,argonaute不具有核酸酶活性,即argonaute与gdna形成特异性识别(即特异性结合)靶基因座而不切割该靶基因座的复合物。ago-gdna复合物的核酸酶活性可使用本领域已知的方法来确定,诸如体外质粒切割测定、t7e1测定、以及测序。参见例如实施例部分中的实验方案。ago-gdna复合物的靶核酸结合活性可使用本领域已知的方法,诸如电泳迁移率变动分析(emsa)来确定。
适用于本申请的方法的argonaute还可具有下列特性中的一者或多者:(1)引导dna在5′端被磷酸化;(2)一旦形成复合物,在低于约50℃的温度下引导dna不与ago蛋白解离;(3)ago核酸酶仅需要单个gdna诱导双链靶dna的双链断裂;(4)ago-gdna复合物可特异性识别和/或切割对gdna的序列具有不超过约3个错配的靶基因座;(5)ago-gdna复合物可特异性识别和/或切割具有至少约60%的gc含量的靶基因座;(6)ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个;以及(7)ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。
具有上述特性的argonaute蛋白可来源于多种生物体的天然存在的argonaute。在一些实施方案中,argonaute来源于原核菌种,诸如古生菌或细菌。在一些实施方案中,ago来源于非嗜热细菌。在一些实施方案中,ago来源于嗜温细菌。在一些实施方案中,ago不来源于嗜热细菌。在一些实施方案中,ago来源于藻类菌种。在一些实施方案中,ago来源于格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌、或解肝磷酯土地杆菌。在一些实施方案中,ago来源于聚球藻属、鞘丝藻属、微囊藻属、伯林盐几何菌、隐红碱线菌、格氏嗜盐碱杆菌、西藏嗜盐碱红菌、嗜热聚球藻、halogeometricumpallidum、解肝磷酯土地杆菌、红细菌目细菌、百脉根中慢生根瘤菌、死海盐盒菌、草根围伯克霍尔德氏菌、burkholderiaambifaria、亚洲嗜盐碱杆菌、或铜绿微囊藻。在一些实施方案中,ago来源于格氏嗜盐碱杆菌,诸如格氏嗜盐碱杆菌sp2。在一些实施方案中,ago来源于铜绿微囊藻,诸如铜绿微囊藻nies843。在一些实施方案中,ago来源于微囊藻属,诸如微囊藻属7806。在一些实施方案中,ago来源于解肝磷酯土地杆菌,诸如解肝磷酯土地杆菌dsm2366。在一些实施方案中,ago不来源于嗜热栖热菌或浑球红细菌(rhodobactersphaeroides)。在一些实施方案中,ago不来源于细长聚球藻。
在一些实施方案中,argonaute是i型原核argonaute。在一些实施方案中,i型原核argonaute携载靶向的dna引导dna。在一些实施方案中,靶向dna核酸的核酸靶向双链dna(dsdna)的一条链,以产生dsdna的切口或断裂。在一些实施方案中,切口或断裂触发宿主dna的修复。在一些实施方案中,宿主dna修复为非同源末端连接(nhej)或同源定向重组(hdr)。在一些实施方案中,i型原核argonaute是长i型原核argonaute。在一些实施方案中,长i型原核argonaute具有n-paz-mid-piwi结构域构造。在一些实施方案中,长i型原核argonaute具有催化活性piwi结构域。在一些实施方案中,长i型原核argonaute具有包含天冬氨酸-谷氨酸-天冬氨酸-天冬氨酸/组氨酸(dedx)基序的催化四联体。在一些实施方案中,催化四联体结合一种或多种二价金属离子,诸如mg2+或mn2+。在一些实施方案中,i型原核argonaute锚定引导dna的5′磷酸基末端。
argonaute蛋白可包含一个或多个结构域。argonaute蛋白可包含选自paz结构域、mid结构域、以及piwi结构域、或它们的任何组合的结构域。argonaute蛋白可包含n-paz-mid-piwi-c的结构域构造。paz结构域可包含寡核苷酸结合折叠区用于固定引导dna的3′末端。从paz结构域释放引导dna的3′末端可有利于argonaute三元复合物转变为切割活性构象。mid结构域可结合gdna的5′磷酸基和第一核苷酸。在一些实施方案中,mid结构域包含5′磷酸基结合位点,该位点具有如图24a所示的kqk基序的3个保守残基中的至少2个或全部3个。在一些实施方案中,kqk基序中的赖氨酸(k)残基被精氨酸(r)置换。在一些实施方案中,kqk基序中的谷氨酰胺(q)残基被天冬酰胺(n)、或带正电的残基诸如赖氨酸(k)或
argonaute蛋白可包含核酸结合结构域。核酸结合结构域可包含接触核酸的区域。核酸结合结构域可结合dna或rna、或dna与rna两者。在一些实施方案中,argonaute蛋白结合dna并切割dna。在一些实施方案中,argonaute蛋白结合单链gdna并切割双链dna。在一些实施方案中,argonaute蛋白结合两个单链gdna并切割双链dna。在一些实施方案中,核酸结合结构域包含paz结构域,其可利用其寡核苷酸结合折叠区来固定所设计的核酸靶向的核酸的3′末端。
argonaute可包括核酸切割结构域,诸如piwi结构域。在一些实施方案中,ago包含含有核酸酶活性位点的piwi结构域。在一些实施方案中,核酸酶活性位点结合二价阳离子。在一些实施方案中,核酸酶活性位点结合两个二价阳离子。例如,第一二价阳离子可引发亲核攻击并活化水分子,并且第二二价阳离子可稳定化过渡态和离去基团。在一些实施方案中,argonaute包含核酸酶结合位点,该位点具有如图24b所示的dde基序的3个保守残基中的至少2个或全部3个。在一些实施方案中,dde基序中的天冬氨酸(d)残基被谷氨酸(e)置换。在一些实施方案中,dde基序中的谷氨酸(e)残基被天冬氨酸(d)置换。在一些实施方案中,核酸酶活性位点还包含一个或多个碱性残基,诸如组氨酸、精氨酸、赖氨酸或它们的组合。组氨酸、精氨酸和/或赖氨酸可在催化和/或切割中发挥作用。在一些实施方案中,核酸酶活性位点包含四个带负电的演化的保守氨基酸,诸如天冬氨酸-谷氨酸-天冬氨酸/组氨酸(dedx,seqidno:157),其形成结合两个二价金属离子(诸如mg2+离子)并将靶核酸切割成带有3′羟基和5′磷酸基的产物的催化四联体。在一些实施方案中,根据ago的类型,该方法在浓度为至少约0.1mm、0.2mm、0.5mm、1mm、2mm、3mm、4mm、5mm、10mm、15mm、20mm的任一种或更大的二价金属离子(诸如mg2+)的存在下进行。argonaute切割靶核酸可发生在一条链的单磷酸二酯键或靶核酸的两条链的至少两个磷酸二酯键处。在一些实施方案中,ago蛋白包含不含dde或dedx(seqidno:157)基序的功能性核酸酶活性位点。在一些实施方案中,ago蛋白不包含功能性核酸酶活性位点。
示例性argonaute及其蛋白质序列示于下表1。在一些实施方案中,argonaute是aaago、afago、baago、bgago、cbago、ccago、cpago、csago、cuago、exago、fpago、haago、hbago、hkago、h1ago、hmago、hpago、ibago、lyago、maago、mfago、migo、mkago、mlago、ago、ngago、npago、ntago、phago、plago、rbago、scago、seago、ssago、syago、tbago、tcago、teago、toago,或它们的功能性衍生物。在一些实施方案中,argonaute为ngago或其功能衍生物。在一些实施方案中,argonaute为phago或其功能衍生物。在一些实施方案中,argonaute为miago或其功能衍生物。在一些实施方案中,argonaute为maago或其功能衍生物。在一些实施方案中,argonaute不为ttago或rsago。在一些实施方案中,argonaute不为seago。
表1.示例性argonaute蛋白序列:
在一些实施方案中,argonaute蛋白包含的序列与表1的野生型argonaute(例如ngago)中的mid结构域、paz结构域和/或piwi结构域有至少约70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或100%的序列同源性。在一些实施方案中,argonaute蛋白包含的序列与表1的野生型argonaute(例如ngago、miago、maago或phago)中的mid结构域、paz结构域和/或piwi结构域有至少约30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或约100%的序列同一性。在一些实施方案中,argonaute蛋白包含的序列与选自seqidno:1-42的序列有至少约70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或100%的序列同源性。在一些实施方案中,argonaute蛋白包含的序列与选自seqidno:1-42的序列有至少约30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或约100%的序列同一性。在一些实施方案中,ago蛋白具有选自seqidno:1-42的序列。在一些实施方案中,argonaute蛋白包含的序列与seqidno:1有至少约70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或100%的序列同源性。在一些实施方案中,argonaute蛋白包含的序列与seqidno:1有至少约30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或约100%的序列同一性。在一些实施方案中,ago蛋白具有seqidno:1。在一些实施方案中,argonaute蛋白包含的序列与seqidno:2有至少约70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或100%的序列同源性。在一些实施方案中,argonaute蛋白包含的序列与seqidno:2有至少约30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或约100%的序列同一性。在一些实施方案中,ago蛋白具有seqidno:2。在一些实施方案中,argonaute蛋白包含的序列与seqidno:11有至少约70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或100%的序列同源性。在一些实施方案中,argonaute蛋白包含的序列与seqidno:11有至少约30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或约100%的序列同一性。在一些实施方案中,ago蛋白具有seqidno:11。在一些实施方案中,argonaute蛋白包含的序列与seqidno:41有至少约70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或100%的序列同源性。在一些实施方案中,argonaute蛋白包含的序列与seqidno:41有至少约30%、40%、50%、60%、70%、75%、80%、85%、90%、95%、97%、99%的任一种、或更大、或约100%的序列同一性。在一些实施方案中,ago蛋白具有seqidno:41。
在一些实施方案中,argonaute是表1中野生型ago蛋白的修饰形式。野生型argonaute的修饰形式可包括使argonaute核酸酶活性降低的氨基酸变化(例如缺失、插入或置换)。例如,修饰的argonaute可具有小于约90%、80%、70%、60%、50%、40%、30%、20%、10%、5%、1%的任一种或更少的野生型argonaute(例如ngago、miago、maago、或phago)的核酸酶活性。argonaute的修饰形式可不具有显著的核酸酶活性。例如,野生型ago的核酸酶活性位点中的dde基序的一个或多个保守残基可突变为例如丙氨酸,以提供不具有核酸酶活性的argonaute。本领域的技术人员将认识到除丙氨酸置换之外的突变也是合适的。在一些实施方案中,可将序列插入argonaute蛋白中以降低其活性。在一些实施方案中,野生型argonaute的修饰形式可具有大于约10%、20%、30%、40%、50%、60%、70%、80%、90%的任一种、或更大的野生型argonaute(例如ngago、miago、maago、或phago)的核酸酶活性。
除非另外指明,否则“argonaute”或“ago”可指对应于野生型argonaute蛋白或其功能性衍生物的多肽,或编码该多肽的多核苷酸。“功能性衍生物”是指这样蛋白质的修饰形式:功能或活性与野生型蛋白质基本上相同,但包括氨基酸序列或化学组成的一种或多种变化,包括但不限于突变(例如缺失、插入、置换等)、非天然氨基酸变体、与其它多肽(例如亲和标签、信号肽等)的融合、与非氨基酸部分(例如染料)的缀合、以及具有其它功能模式的嵌合蛋白。
在一些实施方案中,ago蛋白为融合蛋白。融合蛋白可包含不天然存在于野生型ago中的一种或多种相同的非天然序列。在一些实施方案中,ago蛋白融合于一个或多个亲和标签(诸如ha和flag标签),这可有利于ago蛋白的纯化。在一些实施方案中,ago蛋白融合于荧光蛋白(诸如gfp或rfp),以用于可视化或追踪细胞内的ago蛋白。在一些实施方案中,使ago蛋白融合于自穿透肽,以促进ago-gdna复合物的细胞内递送。在一些实施方案中,使ago蛋白融合于ago的亚细胞定位信号,例如靶向核的核定位信号(nls)、靶向线粒体的线粒体定位信号、靶向叶绿体的叶绿体定位信号、内质网(er)驻留信号等。可使用本领域已知的标准重组方法使融合部分(诸如亲和标签、荧光蛋白、自穿透肽和/或定位信号)融合于蛋白的n-末端或c-末端、或两个末端。
本文还提供了分离的argonaute蛋白以及本文所述的任一种argonaute蛋白用于基因编辑的用途,包括但不限于靶核酸的修饰、靶向编辑细胞内病毒dna、体外靶向编辑dna、以及靶向编辑染色体dna。还提供了任一种argonaute蛋白在制备基因编辑试剂盒或者分析剂或干扰剂中的用途。
编码argonaute蛋白的核酸
本文所述的argonaute蛋白可使用标准重组技术获得,或在宿主细胞(即包含待被ago蛋白修饰的靶核酸的细胞)中重组进行表达。
编码argonaute蛋白的核酸可从表1中所列的任何菌种分离并进行测序。另选地,可基于天然存在的ago序列(诸如seqidno:1-42中的任一种)来设计argonaute编码序列,并且可使用核苷酸合成仪或pcr技术合成具有所设计序列的核酸。在一些实施方案中,ago编码序列诸如通过定点诱变进一步工程化,以表达野生型argonaute蛋白的功能性衍生物或突变型变体。在一些实施方案中,使包含ago编码序列的核酸融合于一种或多种附加组分(诸如信号序列)。在一些实施方案中,对于宿主细胞中ago蛋白的核表达,使核定位序列融合于ago序列的n末端。
在一些实施方案中,编码ago蛋白的核酸经密码子优化以在宿主细胞(诸如真核细胞)中表达。一般来讲,密码子优化是指如下修饰核酸序列以在感兴趣的宿主细胞中增强表达的过程:将天然序列的至少一个密码子(例如约或多于约1个、2个、3个、4个、5个、10个、15个、20个、25个、50个或更多个密码子)用更经常或最经常用于宿主细胞基因的密码子替换,同时保持天然氨基酸序列。各种菌种对特定氨基酸的某些密码子表现出特定偏好。密码子偏好性(生物体间密码子使用的差异)通常与信使rna(mrna)的翻译效率相关联,这继而被认为尤其取决于所翻译密码子的特性以及特定转移rna(trans)分子的可用性。在细胞中占优势的所选择的转移rna通常反映出肽合成中最经常使用的密码子。因此,基因可被定制成基于密码子优化而在给定生物体中进行最佳基因表达。密码子使用表易于例如在“密码子使用数据库(codonusagedatabase)”获得,并且这些表可按多种方式进行调整。参见nakamura,y.等人“codonusagetabulatedfromtheinternationaldnasequencedatabases:statusfortheyear20q0”nuel.acidsres.28:292(2000)。用于密码子优化特定序列以在特定宿主细胞中表达的计算机算法也是可获得的,例如geneforge(aptagen;jacobus,pa)也是可获得的。在一些实施方案中,不需要进行密码子优化。在一些实施方案中,密码子优化是优选的。
在一些实施方案中,将编码ago蛋白的核酸亚克隆到能够在宿主细胞中复制并表达异源多核苷酸的重组载体中。本领域可用且已知的多种载体可用于本发明的目的。合适载体的选择将主要取决于待插入载体中的核酸大小以及待被该载体转化的特定宿主细胞。每种载体包含多种组分,这取决于其功能(异源多核苷酸的扩增或表达,或这两者)及其与其所存在的特定宿主细胞的相容性。在一些实施方案中,提供了用于在原核细胞中表达ago蛋白的载体。在一些实施方案中,提供了用于在真核细胞(诸如哺乳动物细胞)中表达ago蛋白的载体。
“载体”是物质组合物,其包含分离的核酸并且其可用于将分离的核酸递送到细胞的内部。多种载体在本领域中是已知的,包括但不限于线性多核苷酸、与离子或两亲性化合物相关联的多核苷酸、质粒和病毒。
在一些实施方案中,载体为质粒。质粒的示例包括但不限于pgex6p-1和pcdna3.1。在一些实施方案中,质粒是超螺旋质粒。在一些实施方案中,质粒是超纯质粒。如本文所用,“超纯”质粒是指基本上未被内毒素或非病毒微生物污染的质粒制剂,并且质粒为至少约90%、95%、97%、99%的任一种或更多的超螺旋。超纯质粒可使用商业质粒纯化试剂盒进行制备。质粒的超螺旋可利用凝胶电泳来测定。
在一些实施方案中,载体是病毒载体。病毒载体的示例包括但不限于腺病毒载体、腺相关病毒载体、慢病毒载体、逆转录病毒载体、痘苗载体、单纯疱疹病毒载体、以及它们的衍生物。病毒载体技术是本领域公知的,并且描述于例如sambrook等人(2001,molecularcloning:alaboratorymanual,coldspringharborlaboratory,newyork),以及其它病毒学和分子生物学手册。
已经开发出多个基于病毒的系统以使基因转移到哺乳动物细胞中。例如,逆转录酶病毒为基因递送系统提供便利平台。可使用本领域已知的技术将异源核酸插入载体并包装到逆转录病毒颗粒中。然后可分离重组病毒并体外递送至宿主细胞。在一些实施方案中,使用腺病毒载体。在一些实施方案中,使用慢病毒属载体。在一些实施方案中,使用自失活慢病毒载体。
一般来讲,合适的载体包含至少一个生物体内的复制功能起点、启动子序列、适宜的限制性内切核酸酶位点、以及一个或多个选择性标记。载体的附加组分可包括但不限于核糖体结合位点(rbs)、信号序列、以及转录终止序列。一般来讲,包含来源于与宿主细胞相容的菌种的复制子和控制序列的载体与这些宿主结合使用。
在一些实施方案中,核酸可操作地连接至启动子。由多种潜在宿主细胞识别的大量启动子为人们所熟知。在一些实施方案中,启动子是诱导型启动子。在一些实施方案中,启动子是组成型启动子。诱导型启动子是这样的启动子:响应于培养条件变化(例如存在或不存在营养物质或温度变化)而启动在它们控制下的水平提高的编码序列转录。
适合与原核宿主细胞一起使用的启动子包括phoa启动子、-乳糖酶和乳糖启动子系统、碱性磷酸酶启动子、色氨酸(trp)启动子系统、以及杂合启动子,诸如tac启动子。然而,其它已知的细菌启动子也是合适的。用于细菌系统的启动子还将包含可操作地连接于编码ago蛋白的dna的shine-dalgarno(s.d.)序列。
在哺乳动物宿主细胞中由载体转录多肽例如受到启动子的控制,所述启动子获自诸如多瘤病毒、禽痘病毒、腺病毒(诸如腺病毒2)、牛乳头瘤病毒、鸟类肉瘤病毒、细胞巨化病毒、逆转录酶病毒、乙型肝炎病毒并最优选猿猴病毒40(sv40)的病毒的基因组,获自异源哺乳动物启动子例如肌动蛋白启动子或免疫球蛋白启动子,获自热休克启动子,条件是此类启动子与宿主细胞系统相容。
在一些实施方案中,用于在高等真核生物中表达的载体包含增强子序列。许多增强剂序列现已知来自哺乳动物基因(球蛋白、弹性蛋白酶、白蛋白、α-胎蛋白和胰岛素),或来自真核细胞病毒。示例包括复制起点后侧的sv40增强子(bp100-270)、巨细胞病毒早期启动子增强子、复制起点后侧的多瘤增强子、以及腺病毒增强子。增强子可以被剪接到载体中多肽编码序列的5′或3′位置,但优选地位于启动子的5′位点。
用于真核宿主细胞(酵母、真菌、昆虫、植物、动物、人或来自其它多细胞生物体的有核细胞)的表达载体还将包含转录终止和稳定mrna所需的序列。此类序列通常得自真核或病毒dna或cdna的5′(并偶尔为3′)非翻译区。例如,在大多数真核生物的3′端为aataaa序列,它可为向编码序列的3′端添加polya尾巴的信号。这些区域在编码多肽的mrna的非翻译部分中含有转录为聚腺苷酸化片段的核苷酸片段。可将所有这些序列插入真核表达载体中。
在一些实施方案中,编码ago蛋白的核酸是rna。在一些实施方案中,核酸是rna载体。在一些实施方案中,核酸是编码ago蛋白的mrna。在一些实施方案中,编码ago蛋白的mrna包含一种或多种修饰以增强其稳定性、增强其表达和/或降低其免疫原性。在一些实施方案中,mrna包含修饰的主链和/或修饰的核苷酸间键。在一些实施方案中,mrna包含一个或多个硫代磷酸酯键。在一些实施方案中,mrna包含一个或多个修饰的核酸碱基,诸如5-甲基胞嘧啶和/或假尿苷。在一些实施方案中,mrna包含5′帽。
本文还提供了编码本文所述的任何一种argonaute蛋白的核酸和载体(诸如质粒或病毒载体),以及任一种核酸和载体用于ago蛋白表达或用于基因编辑的用途,包括但不限于靶核酸的修饰、靶向编辑细胞内病毒dna、体外靶向编辑dna、以及靶向编辑染色体dna。还提供了任一种核酸和载体在制备基因编辑试剂盒或者分析剂或干扰剂中的用途。
b.引导dna
本文所述的argonaute利用了引导dna,即具有被设计成与待修饰的靶核酸完全互补或基本上互补的序列的单链寡核苷酸dna。在一些实施方案中,ago蛋白需要单个引导dna以诱导双链靶核酸的双链断裂。在一些实施方案中,ago蛋白需要各自靶向双链靶核酸相对链的两个引导dna,以诱导靶核酸的双链断裂。在一些实施方案中,ago蛋白利用靶向靶核酸中的不同序列的多个引导dna。
在一些实施方案中,ago蛋白和引导dna并非天然存在于一起。在一些实施方案中,引导dna包含与宿主细胞的基因组序列的任何部分基本上不杂交的序列。在一些实施方案中,引导dna包含修饰,诸如5′磷酸化,或对核苷酸的碱基或主链部分的修饰,所述修饰不天然存在于宿主细胞中。
在一些实施方案中,ago蛋白不对引导dna中的特定序列或核苷酸有任何偏好。在一些实施方案中,引导dna的5′端被磷酸化。5′磷酸化引导dna可通过化学合成,或通过使用激酶(诸如t4pnk)磷酸化寡核苷酸dna进行制备。在一些实施方案中,5′磷酸化引导dna由细菌细胞内源性地产生。例如,可将质粒转染到细菌细胞中以产生源于质粒的靶向序列的5′磷酸化引导dna。在一些实施方案中,质粒是线性化的质粒。在一些实施方案中,gdna是基本上纯化的,诸如至少约80%、85%、90%、95%、99%的任一种、或更纯。可通过本领域已知的方法纯化引导dna,诸如hplc、凝胶电泳,或使用dna纯化试剂盒。
引导dna的长度可影响ago-gdna复合物的活性(诸如特异性结合和/或核酸酶活性)。在一些实施方案中,引导dna具有约10至约50个核苷酸(“nt”),诸如约10nt至约20nt、约20nt至约30nut、约30nt至约40nt、约40nt至约50nt、约15nt至约30nt、约20nt至约40nt、约15nt至约25nt、或约20nt至约35nt的任一种。在一些实施方案中,引导dna具有约18个、19个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个或30个核苷酸的任一种。在一些实施方案中,引导dna具有约20个核苷酸至约27个核苷酸、或约23个核苷酸至25个核苷酸。在一些实施方案中,其中ago蛋白来源于ngago,引导dna的长度为24个核苷酸。gdna的最佳长度可根据ago蛋白所来源的菌种而变化,并且可由本领域技术人员在体外或细胞内活性测定(例如结合、质粒切割、或报告基因(例如gfp)沉默)中使用不同长度的gdna来确定。
在一些实施方案中,ago-gdna复合物的活性(诸如特异性结合和/或核酸酶活性)对gdna与靶基因座之间的核苷酸错配较敏感。在一些实施方案中,当gdna包含对靶基因座的一个错配时,靶基因座的修饰或切割效率降低至少约10%、20%、30%、40%、50%、60%、70%、80%、90%、95%的任一种或更多。在一些实施方案中,当gdna包含对靶基因座的两个错配(诸如两个连续错配)时,靶基因座的修饰或切割效率降低至少约10%、20%、30%、40%、50%、60%、70%、80%、90%、95%的任一种或更多。在一些实施方案中,当gdna包含对靶基因座的三个错配(诸如三个连续错配)时,靶基因座的修饰或切割效率降低至少约10%、20%、30%、40%、50%、60%、70%、80%、90%、95%的任一种或更多。在一些实施方案中,gdna与靶基因座完全互补。在一些实施方案中,gdna对靶基因座有一个错配。在一些实施方案中,gdna对靶基因座有两个错配。在一些实施方案中,gdna对靶基因座有三个错配。在一些实施方案中,相比于靶基因座的序列,gdna在距5′端的位置8、9、10或11的任一者种中不具有错配。在一些实施方案中,gdna不具有对靶基因座错配的任何三个连续的核苷酸。
在一些实施方案中,引导dna包含修饰的主链和/或修饰的核苷酸间键。在一些实施方案中,引导dna包含一个或多个硫代磷酸酯键。各种盐(例如氯化钾或氯化钠)、混合盐、以及游离酸形式也可包括在内。在一些实施方案中,引导dna包含一个或多个修饰的核酸碱基。
c.ago-gdna复合物
在一些实施方案中,ago蛋白和gdna在细胞中形成复合物。在一些实施方案中,将ago蛋白和gdna提供给体外靶标核酸或者预形成复合物的细胞。一般来讲,ago蛋白和gdna以约1∶1的摩尔比彼此结合。如本文所用,ago蛋白与gdna结合形成的ago-gdna复合物在本文称为gdna“负载”于ago蛋白。预形成的复合物在本文中称为ago“预负载”有gdna。在一些实施方案中,ago-gdna复合物遵循“一种-引导物-忠实性”规则,即ago蛋白不与复合物中结合的gdna解离,或预负载的gdna不与游离未结合的gdna交换。在一些实施方案中,在低于约40℃、45℃、50℃或55℃的任一种温度下,ago蛋白不与预负载的gdna解离。在一些实施方案中,在至少约4小时、8小时、12小时、16小时、24小时的任一种、或更多小时的温育时间段内,在约37℃下,ago蛋白不与预负载的gdna解离或与游离的gdna结合。在一些实施方案中,预形成的ago-gdna复合物不与细胞中不同的gdna交换gdna。
ago-gdna复合物可使用任何合适的方法进行制备。在一些实施方案中,通过使ago蛋白与gdna混合来制备ago-gdna复合物。在一些实施方案中,使ago蛋白和gdna在至少约40℃、45℃、50℃、55℃的任一种或更高的温度下混合,以提供ago-gdna复合物。
在一些实施方案中,通过使ago蛋白在gdna的存在下表达来制备ago-gdna复合物。在一些实施方案中,使编码ago蛋白的核酸(诸如载体)和gdna共转染到宿主细胞中来提供ago-gdna复合物。在一些实施方案中,宿主细胞是不具有内源性5′磷酸化gdna的细胞。在一些实施方案中,宿主细胞是哺乳动物细胞,诸如293t细胞。在一些实施方案中,宿主细胞是细菌细胞,诸如大肠杆菌。在一些实施方案中,使编码ago蛋白的核酸(诸如质粒)和包含gdna的线性化载体共转染到细菌细胞中来提供ago-gdna复合物。在一些实施方案中,使编码ago蛋白的核酸(诸如载体)在gdna的存在下于体外蛋白质翻译系统中表达,以提供ago-gdna复合物。在一些实施方案中,使用本领域已知的蛋白质纯化方法,诸如亲和色谱法和/或尺寸排阻色谱法,从宿主细胞或体外蛋白质翻译系统纯化ago-gdna复合物。在一些实施方案中,ago蛋白包含多肽亲和标签,其可用于纯化来自宿主细胞或体外蛋白质翻译系统的ago-gdna复合物。合适的多肽亲和标签包括但不限于his标签(例如6xhis标签)、血凝素(ha)标签、flag标签、myc标签、gst标签、mbp标签、以及甲壳质结合蛋白标签、钙调蛋白标签、v5标签和链霉抗生物素蛋白结合标签。在一些实施方案中,ago-gdna基本上是纯的。例如,含有预形成的ago-gdna复合物的组合物包含至少约80%、85%、90%、95%、99%的任一种或更多的ago-gdna复合物。
d.靶核酸
本文所公开的方法适用于多种靶核酸。在一些实施方案中,靶核酸是dna。在一些实施方案中,靶核酸是rna,诸如mrna。在一些实施方案中,靶核酸是单链的。在一些实施方案中,靶核酸是双链的。在一些实施方案中,靶核酸包括单链和双链区域两者。在一些实施方案中,靶核酸是线性的。在一些实施方案中,靶核酸是环形的。在一些实施方案中,靶核酸包含一个或多个修饰的核苷酸,诸如甲基化核苷酸、受损核苷酸、或核苷酸类似物。在一些实施方案中,靶核酸未被修饰。
靶核酸可具有任何长度,诸如约至少100bp、200bp、500bp、1000bp、2000bp、5000bp、10kb、20kb、50kb、100kb、200kb、500kb、1mb的任一种或更长。靶核酸还可包含任何序列。在一些实施方案中,靶核酸是富gc靶核酸,诸如具有至少约40%、45%、50%、55%、60%、65%的任一种、或更高的gc含量。在一些实施方案中,靶核酸不是富gc靶核酸。在一些实施方案中,靶核酸具有一种或多种次级结构或高级结构。在一些实施方案中,靶核酸在染色质中不呈凝聚态,使得ago-gdna复合物不可到达靶基因座。
在一些实施方案中,靶核酸存在于细胞中。在一些实施方案中,靶核酸存在于细胞核中。在一些实施方案中,靶核酸对于细胞是内源性的。在一些实施方案中,靶核酸是基因组dna。在一些实施方案中,靶核酸是染色体dna。在一些实施方案中,靶核酸是蛋白质编码基因或其功能区(诸如编码区)、或调控元件(诸如启动子、增强子)、5′或3′非翻译区等。在一些实施方案中,靶核酸是非编码基因,诸如转座子、mirna、trna、核糖体rna、核糖酶或lincrna。在一些实施方案中,靶核酸是质粒。
在一些实施方案中,靶核酸对于细胞是外源性的。在一些实施方案中,靶核酸是病毒核酸,诸如病毒dna或病毒rna。在一些实施方案中,靶核酸是水平转移质粒。在一些实施方案中,靶核酸被整合进细胞的基因组中。在一些实施方案中,靶核酸未整合进细胞的基因组中。在一些实施方案中,靶核酸是细胞中的质粒。在一些实施方案中,靶核酸存在于染色体外阵列中。
在一些实施方案中,靶核酸是分离的核酸,诸如分离的dna或分离的rna。在一些实施方案中,靶核酸存在于无细胞环境中。在一些实施方案中,靶核酸是分离的载体,诸如质粒。在一些实施方案中,靶核酸是超纯质粒。
靶基因座是杂交于gdna的靶核酸片段。在一些实施方案中,ago-gdna复合物切割靶基因座。在一些实施方案中,靶核酸仅具有靶基因座的一个拷贝。在一些实施方案中,靶核酸具有多于一个拷贝,诸如至少约2个、3个、4个、5个、10个、100个拷贝的任一种或更多个靶基因座拷贝。例如,在病毒核酸或细菌的基因组中包含重复序列的靶基因座可被ago-gdna靶定,以抑制或杀伤病毒或细菌。
在一些实施方案中,靶基因座是dna基因座。在一些实施方案中,靶基因座是rna基因座。在一些实施方案中,靶基因座是双链的。在一些实施方案中,靶基因座是单链的。在一些实施方案中,靶基因座是双链dna。
靶基因座可包含任何序列,因为ago蛋白不偏好结合特定序列或序列基序。在一些实施方案中,靶基因座是富gc靶基因座。在一些实施方案中,靶基因座具有至少约40%、50%、60%、70%、80%的任一种或更多的gc含量。在一些实施方案中,靶基因座是非富gc靶核酸中的富gc片段。在一些实施方案中,靶基因座存在于易到达的靶核酸区中。在一些实施方案中,靶基因座处于靶基因的外显子中。在一些实施方案中,靶基因座跨靶基因的外显子-内含子连接区。在一些实施方案中,靶基因座存在于非编码区,诸如基因调控区中。在一些实施方案中,其中靶核酸对于细胞外源,靶基因座包含不存在于细胞基因组的序列。
在一些实施方案中,靶核酸和/或靶基因座包括与信号转导生化途径相关联的序列,例如信号转导生化途径相关的基因或多核苷酸。靶核酸的示例包括疾病相关的基因或多核苷酸。“疾病相关的”基因或多核苷酸是指与非疾病对照的组织或细胞相比,在源于患病组织的细胞中以异常水平或异常形式产生转录或翻译产物的任何基因或多核苷酸。其可能是以异常高的水平表达的基因;其可能是以异常低的水平表达的基因,其中改变的表达与疾病的发生和/或进展有关。疾病相关基因也指有突变或遗传变异的基因,这些突变或遗传变异是疾病病因的直接原因,或者与造成疾病病因的基因处于连锁失衡。转录或翻译产物可为已知或未知的,并且可处于正常或异常水平。这些基因的突变和途径可导致产生影响功能的异常蛋白质或者异常量的蛋白质。在一些实施方案中,靶基因座是疾病相关的基因座。在一些实施方案中,靶基因座包含疾病相关基因的突变或遗传变异。疾病相关的基因和多核苷酸、以及疾病相关的基因座的示例可获自mekusick-nathansinstituteofgeneticmedicine,johnshopkinsuniversity(baltimore,md.)和nationalcenterforbiotechnologyinformation,nationallibraryofmedicine(bethesda,md.),在万维网上可以获得。
本申请设想了产生哺乳动物细胞的同基因细胞系的方法以研究疾病的遗传变异。在一些实施方案中,该方法提供靶基因座处的单核苷酸置换,其可用于研究单核苷酸多态性的影响。本申请还设想了微生物、细胞、植物、动物或合成生物体的基因组修饰,以产生生物、农业和工业可用的产物。该方法可用作用来理解基因组,例如基因敲除或敲入研究的生物研究工具。该方法也可用作靶向细菌感染或病毒感染的特异性菌株的治疗剂。
e.供体dna
在一些实施方案中,该方法包括使靶核酸与包含一个或多个靶核酸的同源序列的供体dna接触。不受任何假设理论的约束,由ago-gdna复合物诱导的靶核酸的双链断裂可引发或刺激细胞中的内源性同源定向重组(hdr)修复途径,该途径将供体dna整合进切割的靶基因座中。在一些实施方案中,供体dna包含5′同源臂、3′同源臂、以及位于5′同源臂和3′同源臂之间的细胞外源序列。在一些实施方案中,同源序列(诸如同源臂)包含与侧接靶核酸的切割位点的序列有至少约80%、85%、90%、95%、99%的任一种、或更大、或100%同一性的序列。在一些实施方案中,同源序列(诸如同源臂)的长度为至少约10个、20个、30个、40个、50个、100个的任一种、或更多个核苷酸。在一些实施方案中,供体dna包含含有靶基因座的靶核酸的置换序列。在一些实施方案中,置换序列与靶核酸的序列的差异在于不超过约10个、5个、4个、3个、2个或1个核苷酸的任一种。
在一些实施方案中,供体dna包含编码选择标记的序列。选择标记可用于选择具有整合进靶基因座的供体dna的细胞。示例性选择标记包括但不限于如下蛋白质:(a)赋予对抗生素或其它毒素,例如氨苄青霉素、新霉素、甲氨蝶呤、四环素、潮霉素和g418的抗性,(b)弥补营养缺陷不足,以及(c)提供不能从复合培养基中获得的关键营养物质。在一些实施方案中,选择方案中使用诸如g418的药物来阻碍生长或杀伤不具有整合进靶基因座的供体dna的细胞。用供体dna成功修饰的那些细胞产生赋予药物抗性的蛋白质,因此在选择方案中得以存活。适用于哺乳动物细胞的选择性标记的示例还包括dhfr、胸苷激酶、金属硫蛋白i和ii(优选灵长类金属硫蛋白基因)、腺苷脱氨酶、鸟氨酸脱羧酶等。
在一些实施方案中,选择标记是允许通过确认报告蛋白的表达而选择的报告蛋白。报告蛋白的示例包括但不限于谷胱甘肽-s-转移酶(gst)、辣根过氧化物酶(hrp)、氯霉素乙酰转移酶(cat)β-半乳糖苷酶、β-葡糖苷酸酶、荧光素酶、绿色荧光蛋白(gfp,例如egfp)、hcred、dsred、青色荧光蛋白(cfp)、黄色荧光蛋白(yfp)、以及自荧光蛋白,包括蓝色荧光蛋白(bfp)。在一些实施方案中,供体dna不包含用于报告蛋白的启动子。因此,报告蛋白仅当供体dna整合进细胞内源性基因的框内时才表达。
供体dna可为任何合适的长度,诸如至少约100bp、200bp、300bp、500bp、1kb、2kb、5kb、10kb的任一种或更长。供体dna可通过化学合成或从模板进行pcr扩增来制备。在一些实施方案中,供体dna基本上是纯化的,诸如至少约80%、85%、90%、95%、99%的任一种或更纯。在一些实施方案中,供体dna存在于载体中。在一些实施方案中,供体dna存在于质粒,诸如超纯质粒或线性化质粒中。可通过本领域已知的方法纯化供体dna,诸如hplc、凝胶电泳,或使用dna纯化试剂盒。
f.细胞
本文所述的方法可用于修饰多种细胞中的靶核酸。在一些实施方案中,细胞是分离细胞。在一些实施方案中,细胞处于细胞培养物中。在一些实施方案中,细胞处于体外。在一些实施方案中,细胞获自活生物体,并维持于细胞培养物中。在一些实施方案中,细胞是单细胞生物体细胞。
在一些实施方案中,细胞是原核细胞。在一些实施方案中,细胞是细菌细胞或来源于细菌细胞。在一些实施方案中,细菌细胞不与ago蛋白所来源的细菌菌种相关。在一些实施方案中,细胞是古细菌细胞或来源于古细菌细胞。在一些实施方案中,细胞是真核细胞。在一些实施方案中,细胞是植物细胞或来源于植物细胞。在一些实施方案中,细胞是真菌细胞或来源于真菌细胞。在一些实施方案中,细胞是动物细胞或来源于动物细胞。在一些实施方案中,细胞是无脊椎动物细胞或来源于无脊椎动物细胞。在一些实施方案中,细胞是脊椎动物细胞或来源于脊椎动物细胞。在一些实施方案中,细胞是哺乳动物细胞或来源于哺乳动物细胞。在一些实施方案中,细胞是人细胞。在一些实施方案中,细胞是斑马鱼细胞。在一些实施方案中,细胞是啮齿动物细胞。在一些实施方案中,细胞经合成制备,有时称为人工细胞。
在一些实施方案中,细胞来源于细胞系。用于组织培养的多种细胞系是本领域已知的。细胞系的示例包括但不限于293t、mf7、k562、hela,以及它们的转基因品种。细胞系可得自本领域技术人员已知的多种来源(参见例如美国典型培养物保藏中心(atcc)(manassus,va.))。在一些实施方案中,用本文所述的一种或多种核酸(诸如ago编码载体和gdna)或ago-gdna复合物转染的细胞用于建立包含一个或多个载体衍生的序列的新细胞系,以建立包含对于靶核酸的修饰的新细胞系。在一些实施方案中,用本文所述的一种或多种核酸(诸如ago编码载体和gdna)或ago-gdna复合物瞬时或非瞬时转染的细胞、或源于此类细胞的细胞系用于评估一种或多种测试化合物。
在一些实施方案中,细胞是初生细胞。例如,初生细胞的培养物可传代0次、1次、2次、4次、5次、10次、15次或更多次。在一些实施方案中,初生细胞通过任何已知的方法从个体收获。例如,白细胞可通过血液成分单采术、白细胞去除疗法、密度梯度分离等收获。诸如皮肤、肌肉、骨髓、脾、肝脏、胰腺、肺、肠、胃等组织的细胞可通过活组织检查收获。合适的溶液可用于分散或悬浮收获的细胞。此类溶液通常可为平衡盐溶液(诸如生理盐水、磷酸盐缓冲盐水(pbs)、hank平衡盐溶液等),其适宜地补充有胎牛血清或其它天然存在的因子、以及可接受的低浓度缓冲液。缓冲液可包括hepes、磷酸盐缓冲液、乳酸盐缓冲液等。细胞可被立即使用,或可将其储存(例如通过冷冻)。可使冷冻细胞解冻,并且能够重新使用。可在冷冻温度下将细胞冷冻于dmso、血清、培养基缓冲液(例如10%dmso、50%血清、40%缓冲培养基)和/或用于保存细胞的一些其它此类常用溶液中。
在一些实施方案中,细胞是免疫细胞,诸如t细胞、自然杀伤细胞和巨噬细胞。在一些实施方案中,细胞是从患者或供体获得的人t细胞。本文提供的方法可用于修饰初级t细胞中的靶核酸以用于免疫疗法。
在一些实施方案中,细胞是干细胞或祖细胞。细胞可包括干细胞(例如,成人干细胞、胚胎干细胞,ips细胞)和祖细胞(例如,心脏祖细胞、神经祖细胞等)。细胞可包括哺乳动物干细胞和祖细胞,包括啮齿动物干细胞、啮齿动物祖细胞、人干细胞、人祖细胞等。
在一些实施方案中,细胞是病变细胞。病变细胞可具有改变的代谢、基因表达和/或形态学特征。病变细胞可为癌细胞、糖尿病细胞和凋亡细胞。病变细胞可为来自患病受治疗者的细胞。示例性疾病可包括血液疾病、癌症、代谢紊乱、眼疾病、器官疾病、肌骨骼病、心脏病等。
在一些实施方案中,细胞不被或基本上不被污染。污染包括内毒素、螫合剂(诸如edta)和微生物,诸如支原体、衣原体、古生菌、原生动物和真菌。在一些实施方案中,ago-gdna对细胞内细菌(诸如支原体)的污染敏感。细胞内细菌可广泛存在,并且不在细胞系中留下存在的可见病征。在实施本文所述的方法之前,应将细胞内细菌小心地排除在细胞之外。在一些实施方案中,将细胞(诸如获自商业来源的细胞系)用一种或多种抗生素诸如青霉素和链霉素处理以除去微生物污染。在一些实施方案中,将细胞培养基热灭活以除去微生物污染。在一些实施方案中,为了将细胞分离并接种到平板中,缓冲液中避免螫合剂如edta。在一些实施方案中,根据细胞类型,将二价离子(诸如mg2+)以至少约0.1mm、0.2mm、0.5mm、1mm、2mm、3mm、4mm、5mm的任一种或更大的浓度补充于细胞。
在一些实施方案中,argonaute诱导核酸(例如基因组dna)的双链断裂或单链断裂。双链断裂可刺激细胞内源性dna修复途径,包括同源定向重组(hdr)、非同源末端连接(nhej)、或可选非同源末端连接(nhej)。nhej可修复切割的靶核酸而无需同源模板。这可导致一个或多个核苷酸在靶基因座处缺失或插入。hdr可采用同源模板诸如供体dna进行。同源模板可包含与侧接靶核酸切割位点的序列同源的序列。在一些情况下,hdr可将外源多核苷酸序列插入切割的靶基因座中。靶dna由于nhej和/或hdr的修饰可导致例如突变、缺失、改变、整合、基因修复、基因置换、基因定位、转基因敲入、基因破坏和/或基因敲除。
在一些实施方案中,使细胞培养物同步,以提高该方法的效率。在一些实施方案中,s和g2期的细胞用于hdr介导的基因编辑。在一些实施方案中,细胞可在任何细胞周期经受该方法。在一些实施方案中,细胞在铺板中显著降低了该方法的功效。在一些实施方案中,该方法以不超过约40%、45%、50%、55%、60%、65%或70%的任一种汇合度应用于细胞培养物。
在一些实施方案中,ago-gdna复合物与细胞中靶基因座的结合募集了一个或多个内源性细胞分子或除dna修复途径之外的途径来修饰靶核酸。例如,在一些实施方案中,ago-gdna复合物募集risc复合物以使mrna沉默。在一些实施方案中,无催化活性的ago可用于沉默靶mrna。在一些实施方案中,ago-gdna复合物的结合阻断一个或多个内源性细胞分子或途径进入靶核酸,从而修饰靶核酸。例如,ago-gdna复合物的结合可阻断内源性转录或翻译机制以降低靶核酸的表达。
g.细胞内递送
在一些实施方案中,该方法包括将一种或多种核酸(例如编码ago蛋白的核酸、gdna、供体dna等)、一种或多种其转录物、和/或预形成的ago-gdna复合物递送到细胞中。示例性的细胞内递送方法包括但不限于:病毒或病毒样试剂;基于化学的转染方法,诸如使用磷酸钙、树枝状体、脂质体、或阳离子聚合物(例如deae-葡聚糖或聚氮丙啶)的那些;非化学方法,诸如显微注射、电穿孔、细胞挤压、声致穿孔、光学转染、穿刺转染(impalefection)、原生质体融合、细菌接合、质粒或转座子递送;基于颗粒的方法,诸如使用基因枪、磁转染或磁体辅助的转染、粒子轰击;以及杂交法,诸如核转染。在一些实施方案中,本申请还提供由此类方法产生的细胞,以及包含此类细胞或由此类细胞产生的生物体(诸如动物、植物或真菌)。
核酸的共转染
在一些实施方案中,该方法包括使编码ago蛋白的核酸(诸如载体)、引导dna、以及任选的供体dna转染到细胞中。在一些实施方案中,使编码ago蛋白的核酸与引导dna同时转染。在一些实施方案中,编码ago蛋白的核酸在引导dna之后转染,诸如在引导dna转染后至少约1小时、2小时、3小时、4小时、5小时、6小时、7小时、8小时的任一种或更多小时。在一些实施方案中,将引导dna转染到携载载体的宿主细胞中,或与编码ago蛋白的核酸稳定整合。在一些实施方案中,将引导dna转染到细胞中两次或多次(例如2次、3次、4次、5次、6次或更多次)。在一些实施方案中,将第一批gdna与编码ago蛋白的核酸同时转染到细胞中,随后在第一转染后至少约2小时、4小时、6小时、8小时、10小时、12小时、16小时、20小时、24小时的任一种或更多小时之后,将一个或多个附加批次的gdna转染到细胞中。在一些实施方案中,使供体dna与编码ago蛋白的核酸和引导dna同时转染。在一些实施方案中,首先将编码ago蛋白的核酸和引导dna转染到细胞中(诸如同时或依次),然后在第一转染后至少约2小时、4小时、6小时、8小时、10小时、12小时、16小时、20小时、24小时的任一种或更多小时之后,将引导dna转染到细胞中。
可使用相同的方法或不同的方法将编码ago蛋白的核酸、引导dna和供体dna转染到细胞中。合适的方法可由本领域的技术人员在多种已知方法中进行选择。可根据转染方法、转染次序、以及转染核酸的性质来选择和调节编码ago蛋白的核酸、引导dna和供体dna的合适剂量。在一些实施方案中,将至少约10ng、50ng、100ng、200ng、300ng、500ng、750ng、1mg的任一种或更多的各编码ago蛋白的核酸、引导dna和任选的供体dna转染到细胞中。在一些实施方案中,将约100ng至500ng的各编码ago蛋白的核酸、引导dna和任选的供体dna转染到细胞中。在一些实施方案中,其中编码ago蛋白的质粒和引导dna同时转染到细胞中,引导dna与质粒之间的重量比为至少约1∶1、1∶2、1∶3、1∶4、或1∶5。在一些实施方案中,转染进细胞中的引导dna与编码ago蛋白的核酸(诸如载体)之间的摩尔比为至少约10∶1、20∶1、50∶1、100∶1、150∶1、200∶1、300∶1、500∶1、750∶1、1000∶1的任一种或更大。在一些实施方案中,转染进细胞中的引导dna与供体dna之间的摩尔比为至少约1∶5、1∶4、1∶3、1∶2、1∶1、2∶1、3∶1、4∶1、5∶1的任一种或更大。在一些实施方案中,转染进细胞中的供体dna与编码ago蛋白的核酸(诸如载体)之间的摩尔比为至少约10∶1、20∶1、50∶1、100∶1、150∶1、200∶1、300∶1、500∶1、750∶1、1000∶1的任一种或更大。
核酸的非病毒递送方法包括脂质体转染、核转染、显微注射、基因枪、病毒体、脂质体、免疫脂质体、聚阳离子或类脂、核酸缀合物、裸dna、人工病毒体、以及dna的试剂增强性吸收。脂质体转染描述于例如美国专利no.5,049,386、4,946,787和4,897,355中,并且脂转染试剂有商业出售(例如transfectaminetm和
用于核酸递送的常规基于病毒的系统包括逆转录病毒、慢病毒、腺病毒、腺相关和单纯疱疹病毒载体。可能采用逆转录病毒、慢病毒、以及腺相关的病毒方法实现的宿主基因组的整合,这通常导致所插入转基因长期表达。另外,在许多不同的细胞类型中观察到较高的转导效率。逆转录病毒的趋性可通过引入外来包膜蛋白进行改变,从而扩增靶细胞的潜在靶群体。慢病毒载体是能够转导或感染非分裂细胞并通常产生高病毒效价的逆转录病毒载体。逆转录病毒载体由具有包装最多至6kb-10kb外源序列的能力的顺式作用长末端重复序列构成。最小顺式作用ltr足以复制和包装载体,其然后用于将核酸整合进靶细胞中以提供永久的转基因表达。广泛使用的逆转录病毒载体包括基于小鼠白血病病毒(mulv)、长臂猿白血病病毒(galv)、类人猿免疫缺陷病毒(siv)、人免疫缺陷病毒(hiv)、以及它们的组合的那些。在优选瞬时表达的应用中,可使用基于腺病毒的系统。基于腺病毒的载体在许多细胞类型中具有极高的转导效率并且不需要细胞分裂。在此类载体情况下,获得较高的滴度和表达水平。该载体可在相对简单的系统中大量产生。
包装细胞通常用于形成能够感染宿主细胞的病毒颗粒。此类细胞包括包装腺病毒的293t细胞,以及包装逆转录病毒的ψ2细胞或pa317细胞。病毒载体通常通过产生使核酸载体包装到病毒颗粒中的细胞系而生成。载体通常包含用于包装并随后整合进宿主中所需的最小病毒序列,被待表达多核苷酸的表达盒替代的其它病毒序列。包装细胞系通常反式提供缺失的病毒功能。例如,用于基因疗法的aav载体通常仅具有来自于包装和整合进宿主基因组所需的aav基因组的itr序列。病毒dna被包装到细胞系中,该细胞系包含编码其它aav基因(即rep和cap)但缺乏itr序列的辅助质粒。细胞系也可被腺病毒作为辅助病毒感染。辅助病毒促进aav载体的复制和辅助质粒的aav基因的表达。辅助质粒由于缺少itr序列而不以显著量包装。腺病毒的污染可通过例如相比于aav而言腺病毒对其更为敏感的热处理降低。
ago-gdna复合物的递送
在一些实施方案中,该方法包括将预形成的ago-gdna复合物递送到细胞中。本领域已知的用于蛋白质或肽递送的任何方法可用于递送ago-gdna复合物。在一些实施方案中,复合物中gdna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,该方法还包括在将ago-gdna复合物递送至细胞后至少约2小时、4小时、6小时、8小时、10小时、12小时、16小时、20小时、24小时的任一种、或更多小时,使供体dna转染到细胞中。在一些实施方案中,该方法包括将预形成的ago-gdna复合物同时(诸如以同一组合物)递送到细胞中。
用于细胞内递送蛋白质或蛋白质复合物(诸如预形成的ago-gdna复合物)的方法包括但不限于机械法,诸如使用微流体装置对细胞显微注射、电穿孔和机械变形;基于载体的方法,诸如细胞穿透肽(cpp)、病毒样颗粒、超电荷蛋白质(superchargedproteins)、纳米载体、基于超分子载体的递送系统、以及纳米颗粒稳定的纳米胶囊。参见例如fu等人bioconjugatechem.2014,25,1602-1608。一些机械方法诸如显微注射和电穿孔可为侵入式的,并且通量较低。在一些实施方案中,通过穿过细胞膜插入蛋白质,同时使细胞通过微流系统统诸如cell
对于基于载体的递送,在一些实施方案中,ago蛋白例如通过共价连接或重组融合于载体而融合到载体上。在一些实施方案中,ago-gdna复合物经由一个或多个细胞穿透肽(cpp)递送至细胞。示例性cpp包括但不限于tat肽、聚精氨酸肽(诸如r9肽)、pep-1、穿膜肽、nrtps、以及它们的衍生物。在一些实施方案中,通过重组方法,或通过ago蛋白的翻译后修饰,使cpp融合于ago蛋白的n端或c末端。在一些实施方案中,将一种或多种ph和温度诱导的调节剂、合成胞内体裂解剂和/或光诱导的物理破碎应用于细胞,以利于细胞穿透肽所递送的ago-gdna复合物胞内体逃逸。
在一些实施方案中,ago-gdna复合物经由超电荷蛋白质递送至细胞。超电荷蛋白质是一类具有异常高正或负净理论电荷(通常每千道尔顿分子量的净电荷单位>1)的工程化或天然存在的蛋白质。例如,超电荷gfp可用于将大分子递送到细胞中。参见例如cronican等人chem.biol.2011,18:833-838。在一些实施方案中,超电荷蛋白质融合于ago蛋白的n-末端或c-末端。在一些实施方案中,ago-gdna复合物经由共价连接的纳米载体(诸如磁纳米粒子或介孔二氧化硅纳米粒子)递送至细胞。参见例如donae和burda.adv.drugdeliveryrev.,2013,65:607-621。
在一些实施方案中,ago-gdna与载体相缔合以提供超分子递送系统。在一些实施方案中,ago-gdna复合物经由病毒样颗粒(vlp)递送至细胞。vlp通过病毒衣壳蛋白的自组装形成,其大小和构象类似于完整的感染性病毒体,但具有非病毒特性,包括非复制、非病原性、以及无基因组(genomeless)。参见例如muratori等人methodsmol.biol.2010,614:111-124;以及kaczmarczyk等人pnas2011,108:16998-17003。在一些实施方案中,ago-gdna复合物经由脂质体载体(诸如基于赖氨酸的阳离子脂质体)递送至细胞。脂质体包封的ago-gdna复合物的脂质体可使用本领域已知的方法制备,包括例如无解冻循环过程。在一些实施方案中,ago-gdna复合物经由脂质复合物递送至细胞。脂质复合物可包含表面活性剂、蛋白质、脂质、聚合物、或这些材料的组合,并且包括固体脂质颗粒、油性悬浮液、亚微米脂质乳液、脂质植入物、脂质微泡、反脂质胶团、脂质微管、脂质球和脂质微圆柱体。可商购获得的阳离子脂质试剂诸如
h.细胞评估和选择
在一些实施方案中,在将编码ago蛋白的核酸和gdna、或ago-gdna复合物、以及任选的供体dna递送到细胞中之后,将细胞培养至少约8小时、12小时、24小时、48小时、60小时、3天、4天、6天的任一种、或更多天,诸如约48小时至60天。在一些实施方案中,允许细胞在评估和/或选择之前生长至不超过约95%、90%、85%、或80%的汇合度的任一种。在一些实施方案中,细胞基于一个或多个特征进行选择,包括但不限于选择标记(例如抗生素抗性蛋白、荧光标签等)的表达、靶核酸的表达水平、细胞的表型变化、或靶核酸的序列(例如靶基因座的pcr扩增子)。在一些实施方案中,分离出靶核酸成功修饰的单个克隆。
在一些实施方案中,对细胞的表型变化进行评估。在一些实施方案中,其中外源基因被敲入至细胞,评估与外源基因相关联的表型变化,以选择外源基因的成功整合的细胞。在一些实施方案中,其中突变引入至基因,评估与突变或基因相关联的表型变化。例如,如果基因参与发育,则可评估细胞或与突变相关联的细胞所来源的生物体中的发育缺陷,以鉴别成功引入突变的细胞。适用于筛选的其它示例性表型变化包括细胞的生长速率、细胞周期进程和代谢表型。表型变化可使用已知针对靶核酸及其相关表型选择的测定法来确定,包括例如显微镜法。
在一些实施方案中,对靶核酸的表达水平进行评估。基因的表达水平可以rna水平或蛋白质水平进行测定。许多方法在本领域已知用于评估表达水平,包括但不限于用于蛋白质水平的蛋白质印记杂交和免疫染色、以及用于rna水平的定量rt-pcr、rnaseq和原位杂交。在一些实施方案中,表达水平降低至少约10%、20%、30%、40%、50%、60%、70%、80%、90%的任一种、或更大。
在一些实施方案中,其中供体dna包含选择标记,对选择标记的表达水平进行评估。在一些实施方案中,其中选择标记为抗生素抗性基因,在将ago-gdna复合物或者编码ago的基因与gdna递送到细胞中之后例如约48小时至60小时,将适当的抗生素补充于细胞,以选择出供体dna成功整合进靶核酸的细胞。在一些实施方案中,其中选择标记为荧光蛋白(例如mrfp、egfp等),通过荧光显微法或通过荧光辅助的细胞分类(facs)评估选择标记的表达。
在一些实施方案中,对靶基因座进行扩增并评估,以选择出具有期望的突变、或外源序列敲入的细胞。pcr引物可被设计成用于在靶核酸的例如靶基因座与供体dna的插入序列之间的接头处扩增修饰区,以提供扩增子进行分析。在一些实施方案中,可通过凝胶电泳、限制性消化,或通过测序(诸如桑格测序)来分析pcr扩增子,以确认靶基因座如所设计的那样修饰。
iii.组合物、递送系统和试剂盒
本专利申请还提供了用于实施本文所述任一种方法的组合物、递送系统、试剂盒。
在一些实施方案中,提供了一种组合物,该组合物包含含有ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的复合物,其中复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。在一些实施方案中,ago蛋白包括核定位信号(nls)。
在一些实施方案中,提供了一种组合物,该组合物包含含有ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的复合物,其中ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)序列同源性的氨基酸序列。在一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。在一些实施方案中,复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。
在一些实施方案中,提供了一种组合物,该组合物包含含有ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的复合物,其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)序列同源性的氨基酸序列。在一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。在一些实施方案中,复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。
在一些实施方案中,提供了一种递送系统,该递送系统包含含有ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的复合物以及适于细胞内递送该复合物的载体,其中复合物能够在约10℃至约60℃(诸如37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,载体选自细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物和纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。在一些实施方案中,ago蛋白包括核定位信号(nls)。
在一些实施方案中,提供了一种递送系统,该递送系统包含含有ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的复合物以及适于细胞内递送该复合物的载体,其中ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)序列同源性的氨基酸序列。在一些实施方案中,载体选自细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物和纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。在一些实施方案中,复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。
在一些实施方案中,提供了一种递送系统,该递送系统包含含有ago蛋白和单链引导dna(诸如5′磷酸化单链引导dna)的复合物以及适于细胞内递送该复合物的载体,其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)序列同源性的氨基酸序列。在一些实施方案中,载体选自细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物和纳米颗粒稳定的纳米胶囊。在一些实施方案中,使ago蛋白融合于细胞穿透肽。在一些实施方案中,引导dna和ago蛋白不天然存在于同一生物体中。在一些实施方案中,复合物能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。
在一些实施方案中,提供了一种试剂盒,该试剂盒包含编码ago蛋白的核酸和单链引导dna(诸如5′磷酸化单链引导dna),其中ago蛋白和引导dna形成能够在约10℃至约60℃的温度下特异性识别靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,编码ago蛋白的核酸对感兴趣的菌种是密码子优化的。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,试剂盒还包含供体dna。
在一些实施方案中,提供了一种试剂盒,该试剂盒包含编码ago蛋白的核酸和单链引导dna(诸如5′磷酸化单链引导dna),其中ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。在一些实施方案中,ago蛋白包含与选自seqidno:1-42的序列具有至少约80%(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)序列同源性的氨基酸序列。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,编码ago蛋白的核酸对感兴趣的菌种是密码子优化的。在一些实施方案中,ago蛋白和引导dna形成能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,试剂盒还包含供体dna。
在一些实施方案中,提供了一种试剂盒,还试剂盒包含编码ago蛋白的核酸和单链引导dna(诸如5′磷酸化单链引导dna),其中ago蛋白来源于格氏嗜盐碱杆菌。在一些实施方案中,ago蛋白包含与seqidno:1具有至少约80%(诸如至少约85%、90%、95%、98%、99%的任一种或更大的序列同源性,或约100%的序列同一性)序列同源性的氨基酸序列。在一些实施方案中,编码ago蛋白的核酸可操作地连接至启动子。在一些实施方案中,编码ago蛋白的核酸存在于载体,诸如病毒载体中。在一些实施方案中,载体是超纯质粒。在一些实施方案中,编码ago蛋白的核酸是mrna。在一些实施方案中,编码ago蛋白的核酸对感兴趣的菌种是密码子优化的。在一些实施方案中,ago蛋白和引导dna形成能够在约10℃至约60℃(诸如约37℃)的温度下特异性识别靶基因座的复合物,并且其中靶基因座包含与引导dna的序列互补的序列。在一些实施方案中,ago蛋白能够切割靶基因座。在一些实施方案中,其中靶基因座是双链dna,ago蛋白能够诱导靶基因座的双链断裂。在一些实施方案中,ago蛋白不能够切割靶基因座。在一些实施方案中,在低于约50℃的温度下引导dna不与ago蛋白解离。在一些实施方案中,引导dna的长度为约10个至约50个核苷酸(诸如约20个至约30个核苷酸)。在一些实施方案中,靶基因座的序列包含对引导dna的序列的不超过约3个错配(诸如无错配)。在一些实施方案中,靶基因座具有至少约60%的gc含量。在一些实施方案中,引导dna与ago蛋白之间的摩尔比为至少约1∶1。在一些实施方案中,ago蛋白包括核定位信号(nls)。在一些实施方案中,试剂盒还包含供体dna。
还提供了包含本文所述任一种组合物或递送系统的细胞、试剂盒和制品。
在一些实施方案中,试剂盒包含一种或多种试剂用于本文所述的任一种方法。试剂可提供于任何合适的容器中。例如,试剂盒可提供一种或多种反应或储存缓冲液。试剂能够以可用于特定测定法的形式,或以需要在使用前添加一种或多种其它组分的形式(例如浓缩或冻干形式)提供。缓冲液可为任何缓冲液,包括但不限于碳酸钠缓冲液、碳酸氢钠缓冲液、硼酸盐缓冲液、tris缓冲液、mops缓冲液、hepes缓冲液,以及它们的组合。在一些实施方案中,缓冲液是碱性缓冲液。在一些实施方案中,缓冲剂的ph为约7至约10。在一些实施方案中,试剂盒包含供体dna。
在一些实施方案中,试剂盒还包括用于实施本文所述的任一种方法的说明书。本文所述试剂盒可用于修饰靶核酸、基因组编辑、在靶基因座处引入突变(例如插入缺失、移码、敲除或敲入、以及置换)、改变细胞表型,作为分析物或干扰剂。
示方案性实施方案
以下示例性实施方案旨在仅对本发明进行举例说明,并因此不应被视为以任何方式限制本发明。
实施方案1.在一些实施方案中,提供了一种修饰靶核酸的方法,包括在约10℃至约60℃的温度下使靶核酸与argonaute(ago)蛋白和单链引导dna接触,其中所述ago蛋白和所述引导dna形成特异性识别所述靶核酸中的靶基因座的复合物,并且其中所述靶基因座包含与所述引导dna的序列互补的序列。
实施方案2:在根据实施方案1所述的另一些实施方案中,所述ago蛋白切割所述靶基因座。
实施方案3:在根据实施方案2所述的另一些实施方案中,其中所述靶基因座是双链dna,所述ago蛋白诱导所述靶基因座的双链断裂。
实施方案4:在根据实施方案1-3中任一项所述的另一些实施方案中,所述温度为约37℃。
实施方案5:在根据实施方案1-4中任一项所述的另一些实施方案中,所述ago蛋白和所述引导dna存在于预形成的复合物中。
实施方案6:在根据实施方案5所述的另一些实施方案中,在低于约50℃的温度下所述引导dna不与所述ago蛋白解离。
实施方案7:在根据实施方案1-6中任一项所述的另一些实施方案中,所述靶基因座的序列包含对所述引导dna的序列的不超过约3个错配。
实施方案8:在根据实施方案1-7中任一项所述的另一些实施方案中,所述靶基因座具有至少约60%的gc含量。
实施方案9:在根据实施方案1-8中任一项所述的另一些实施方案中,所述ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。
实施方案10:在根据实施方案1-9中任一项所述的另一些实施方案中,所述ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。
实施方案11:在根据实施方案1-10中任一项所述的另一些实施方案中,所述ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。
实施方案12:在实施方案11所述的另一些实施方案中,所述ago蛋白来源于格氏嗜盐碱杆菌。
实施方案13:在实施方案11所述的另一些实施方案中,所述ago蛋白来源于解肝磷酯土地杆菌。
实施方案14:在实施方案11所述的另一些实施方案中,所述ago蛋白来源于微囊藻属。
实施方案15:在实施方案11所述的另一些实施方案中,所述ago蛋白来源于铜绿微囊藻。
实施方案16:在根据实施方案1-11中任一项所述的另一些实施方案中,所述ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性的氨基酸序列。
实施方案17:在实施方案16所述的另一些实施方案中,所述ago蛋白包含与seqidno:1具有至少约80%序列同源性的氨基酸序列。
实施方案18:在实施方案16所述的另一些实施方案中,所述ago蛋白包含与seqidno:2具有至少约80%序列同源性的氨基酸序列。
实施方案19:在实施方案16所述的另一些实施方案中,所述ago蛋白包含与seqidno:11具有至少约80%序列同源性的氨基酸序列。
实施方案20:在实施方案16所述的另一些实施方案中,所述ago蛋白包含与seqidno:41具有至少约80%序列同源性的氨基酸序列。
实施方案21:在根据实施方案16-20中任一项所述的另一些实施方案中,所述至少约80%的序列同源性是100%的序列同一性。
实施方案22:在根据实施方案1-21中任一项所述的另一些实施方案中,所述引导dna在5′端被磷酸化。
实施方案23:在根据实施方案1-22中任一项所述的另一些实施方案中,所述引导dna的长度为约10个至约50个核苷酸。
实施方案24:在根据实施方案1-23中任一项所述的另一些实施方案中,所述接触在二价金属离子的存在下进行。
实施方案25:在实施方案24所述的另一些实施方案中,所述二价金属离子的浓度为至少约0.1mm。
实施方案26:在根据实施方案1-25中任一项所述的另一些实施方案中,所述靶核酸是分离的dna。
实施方案27:在根据实施方案1-25中任一项所述的另一些实施方案中,所述靶核酸存在于细胞中。
实施方案28:在根据实施方案27所述的另一些实施方案中,所述方法包括用所述引导dna和编码所述ago蛋白的核酸转染所述细胞。
实施方案29:在根据实施方案28所述的另一些实施方案中,使所述引导dna和编码所述ago蛋白的所述核酸同时转染到所述细胞中。
实施方案30:在根据实施方案28所述的另一些实施方案中,所述引导dna在编码所述ago蛋白的所述核酸之前转染到所述细胞中。
实施方案31:在根据实施方案28-30中任一项所述的另一些实施方案中,用所述引导dna转染所述细胞至少两次。
实施方案32:在根据实施方案28-31中任一项所述的另一些实施方案中,所述引导dna与编码所述ago蛋白的所述核酸的摩尔比为至少约100∶1。
实施方案33:在根据实施方案28-32中任一项所述的另一些实施方案中,编码所述ago蛋白的所述核酸可操作地连接至启动子。
实施方案34:在根据实施方案28-33中任一项所述的另一些实施方案中,编码所述ago蛋白的所述核酸存在于载体中。
实施方案35:在实施方案34所述的另一些实施方案中,所述载体是病毒载体。
实施方案36:在实施方案34或35所述的另一些实施方案中,所述载体是超纯质粒。
实施方案37:在根据实施方案28-32中任一项所述的另一些实施方案中,编码所述ago蛋白的所述核酸是mrna。
实施方案38:在根据实施方案28-37中任一项所述的另一些实施方案中,编码所述ago蛋白的所述核酸对细胞所来源的生物体是密码子优化的。
实施方案39:在根据实施方案27所述的另一些实施方案中,所述方法包括将包含所述ago蛋白和所述引导dna的预形成的复合物递送到所述细胞中。
实施方案40:在根据实施方案39所述的另一些实施方案中,所述预形成的复合物通过电穿孔、显微注射或机械变形经由微流体通道递送到所述细胞中。
实施方案41:在根据实施方案39所述的另一些实施方案中,所述预形成的复合物经由选自以下的载体递送到所述细胞中:细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物、以及纳米颗粒稳定的纳米胶囊。
实施方案42:在实施方案41所述的另一些实施方案中,使所述ago蛋白融合于所述细胞穿透肽。
实施方案43:在根据实施方案27-42中任一项所述的另一些实施方案中,所述方法包括用一种或多种抗生素处理所述细胞。
实施方案44:在根据实施方案27-43中任一项所述的另一些实施方案中,所述ago蛋白包括核定位信号(nls)。
实施方案45:在根据实施方案27-44中任一项所述的另一些实施方案中,所述靶核酸对于所述细胞是内源性的。
实施方案46:在实施方案45所述的另一些实施方案中,所述靶核酸是基因组dna。
实施方案47:在根据实施方案27-44中任一项所述的另一些实施方案中,所述靶核酸对于所述细胞是外源性的。
实施方案48:在实施方案47所述的另一些实施方案中,所述靶核酸是病毒dna。
实施方案49:在实施方案47或48所述的另一些实施方案中,所述靶核酸被整合进所述细胞的基因组中。
实施方案50:在实施方案47或48所述的另一些实施方案中,所述靶核酸未整合进所述细胞的基因组中。
实施方案51:在根据实施方案27-50中任一项所述的另一些实施方案中,所述细胞是原核细胞。
实施方案52:在根据实施方案27-50中任一项所述的另一些实施方案中,所述细胞是真核细胞。
实施方案53:在实施方案52所述的另一些实施方案中,所述细胞是哺乳动物细胞。
实施方案54:在实施方案53所述的另一些实施方案中,所述细胞是人细胞。
实施方案55:在实施方案52所述的另一些实施方案中,所述细胞是酵母细胞、真菌细胞或植物细胞。
实施方案56:在根据实施方案52-55中任一项所述的另一些实施方案中,所述细胞来源于细胞系。
实施方案57:在根据实施方案52-55中任一项所述的另一些实施方案中,所述细胞是初生细胞。
实施方案58:在实施方案57所述的另一些实施方案中,所述细胞是免疫细胞。
实施方案59:在根据实施方案27-58中任一项所述的另一些实施方案中,所述细胞未被非病毒微生物污染。
实施方案60:在根据实施方案1-59中任一项所述的另一些实施方案中,所述修饰包括所述靶核酸的位点特异性切割。
实施方案61:在根据实施方案27-59中任一项所述的另一些实施方案中,所述修饰包括在所述靶基因座处引入选自插入、缺失和移码突变的突变。
实施方案62:在根据实施方案27-59中任一项所述的另一些实施方案中,所述方法还包括在允许供体dna在所述靶基因座处整合的条件下,使所述靶核酸与包含与所述靶基因座的序列同源的序列的所述供体dna接触。
实施方案63:在实施方案62所述的另一些实施方案中,所述供体dna编码选择标记。
实施方案64:在实施方案63所述的另一些实施方案中,所述选择标记是报告蛋白。
实施方案65:在实施方案63或64所述的另一些实施方案中,所述方法还包括评估所述细胞对所述选择标记的表达。
实施方案66:在根据实施方案27-65中任一项所述的另一些实施方案中,所述修饰包括诱导所述细胞的表型变化。
实施方案67:在实施方案66所述的另一些实施方案中,所述方法还包括评估所述细胞的表型变化。
实施方案68:在根据实施方案27-67中任一项所述的另一些实施方案中,所述方法还包括在所述修饰之后对所述靶核酸测序。
实施方案69:在根据实施方案27-68中任一项所述的另一些实施方案中,所述修饰包括改变所述靶核酸的表达。
实施方案70:在根据实施方案27-69中任一项所述的另一些实施方案中,所述修饰包括在所述靶基因座处引入基因敲除突变。
实施方案71:在根据实施方案62-70中任一项所述的另一些实施方案中,所述修饰包括在所述靶基因座处敲入外源序列,其中所述供体dna包含所述外源序列。
实施方案72:在根据实施方案62-70中任一项所述的另一些实施方案中,所述修饰包括在所述靶基因座处引入置换突变,其中所述供体dna包含所述置换突变。
实施方案73:在实施方案72所述的另一些实施方案中,所述置换突变是单核苷酸置换。
实施方案74:在根据实施方案27-73中任一项所述的另一些实施方案中,所述靶基因座是疾病相关的基因座。
实施方案75:在一些实施方案中,提供了一种组合物,所述组合物包含含有ago蛋白和单链引导dna的复合物,其中所述复合物能够在约10℃至约60℃的温度下特异性识别靶基因座,并且其中所述靶基因座包含与所述引导dna的序列互补的序列。
实施方案76:在一些实施方案中,提供了一种递送系统,所述递送系统包含含有ago蛋白和单链引导dna的复合物以及适于细胞内递送所述复合物的载体,其中所述复合物能够在约10℃至约60℃的温度下特异性识别靶基因座,并且其中所述靶基因座包含与所述引导dna的序列互补的序列。
实施方案77:在根据实施方案76所述的另一些实施方案中,所述载体选自细胞穿透肽、病毒样颗粒、纳米载体、脂质体、聚合物和纳米颗粒稳定的纳米胶囊。
实施方案78:在实施方案76或77所述的另一些实施方案中,使所述ago蛋白融合于所述细胞穿透肽。
实施方案79:在根据实施方案75-78中任一项所述的另一些实施方案中,所述引导dna与所述ago蛋白之间的摩尔比为至少约1∶1。
实施方案80:在一些实施方案中,提供了一种试剂盒,所述试剂盒包含编码ago蛋白的核酸和单链引导dna,其中所述ago蛋白和所述引导dna形成能够在约10℃至约60℃的温度下特异性识别靶基因座的复合物,并且其中所述靶基因座包含与所述引导dna的序列互补的序列。
实施方案81:在根据实施方案80所述的另一些实施方案中,编码所述ago蛋白的所述核酸可操作地连接至启动子。
实施方案82:在根据实施方案80或81所述的另一些实施方案中,编码所述ago蛋白的所述核酸存在于载体中。
实施方案83:在实施方案82所述的另一些实施方案中,所述载体是病毒载体。
实施方案84:在实施方案82或83所述的另一些实施方案中,所述载体是超纯质粒。
实施方案85:在根据实施方案84所述的另一些实施方案中,编码所述ago蛋白的所述核酸是mrna。
实施方案86:在根据实施方案80-85中任一项所述的另一些实施方案中,编码所述ago蛋白的所述核酸对感兴趣的生物体是密码子优化的。
实施方案87:在根据实施方案75-86中任一项所述的另一些实施方案中,所述ago蛋白能够切割所述靶基因座。
实施方案88:在根据实施方案75-87中任一项所述的另一些实施方案中,其中所述靶基因座是双链dna,所述ago蛋白能够诱导所述靶基因座的双链断裂。
实施方案89:在根据实施方案75-88中任一项所述的另一些实施方案中,所述温度为约37℃。
实施方案90:在根据实施方案75-89中任一项所述的另一些实施方案中,在低于约50℃的温度下所述引导dna不与所述ago蛋白解离。
实施方案91:在根据实施方案75-90中任一项所述的另一些实施方案中,所述引导dna和所述ago蛋白不天然存在于同一生物体中。
实施方案92:在根据实施方案75-91中任一项所述的另一些实施方案中,所述引导dna在5′端被磷酸化。
实施方案93:在根据实施方案75-92中任一项所述的另一些实施方案中,引导dna的长度为约10个至约50个核苷酸。
实施方案94:在根据实施方案75-93中任一项所述的另一些实施方案中,所述靶基因座的序列包含对所述引导dna的序列的不超过约3个错配。
实施方案95:在根据实施方案75-94中任一项所述的另一些实施方案中,所述靶基因座具有至少约60%的gc含量。
实施方案96:在根据实施方案75-95中任一项所述的另一些实施方案中,所述ago蛋白在5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个。
实施方案97:在根据实施方案75-96中任一项所述的另一些实施方案中,所述ago蛋白在核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个。
实施方案98:在根据实施方案75-97中任一项所述的另一些实施方案中,所述ago蛋白来源于选自以下的生物体:格氏嗜盐碱杆菌、铜绿微囊藻、halogeometricumpallidum、亚洲嗜盐碱杆菌、西藏嗜盐碱红菌、隐红碱线菌、伯林盐几何菌、thermococcusbarophilus、嗜热聚球藻、嗜冷嗜盐菌、微囊藻属、聚球藻属、梭菌属、产气荚膜梭菌、煎盘梭菌、梭菌属、intestinibacterbartlettii、嗜热古菌、盐杆菌属、methanocaldococcusfervens、浅黄假单胞菌、thermogladiuscellulolyticus、固氮弧菌属物种、深海超嗜热古菌、坎式甲烷火菌、细长聚球藻、好热黄无氧芽孢菌、微小杆菌属、鞘丝藻属、丁酸梭菌、halorubrumkocurii、burkholderiaambifaria、草根围伯克霍尔德氏菌、死海盐盒菌、百脉根中慢生根瘤菌、红细菌目细菌和解肝磷酯土地杆菌。
实施方案99:在实施方案98所述的另一些实施方案中,所述ago蛋白来源于格氏嗜盐碱杆菌。
实施方案100:在实施方案98所述的另一些实施方案中,所述ago蛋白来源于解肝磷酯土地杆菌。
实施方案101:在实施方案98所述的另一些实施方案中,所述ago蛋白来源于微囊藻属。
实施方案102:在实施方案98所述的另一些实施方案中,所述ago蛋白来源于铜绿微囊藻。
实施方案103:在根据实施方案75-98中任一项所述的另一些实施方案中,所述ago蛋白包含与选自seqidno:1-42的序列具有至少约80%序列同源性的氨基酸序列。
实施方案104:在实施方案103所述的另一些实施方案中,所述ago蛋白包含与seqidno:1具有至少约80%序列同源性的氨基酸序列。
实施方案105:在实施方案103所述的另一些实施方案中,所述ago蛋白包含与seqidno:2具有至少约80%序列同源性的氨基酸序列。
实施方案106:在实施方案103所述的另一些实施方案中,所述ago蛋白包含与seqidno:11具有至少约80%序列同源性的氨基酸序列。
实施方案107:在实施方案103所述的另一些实施方案中,所述ago蛋白包含与seqidno:41具有至少约80%序列同源性的氨基酸序列。
实施方案108:在根据实施方案103-107中任一项所述的另一些实施方案中,所述至少约80%的序列同源性是100%的序列同一性。
实施方案109:在根据实施方案75-108中任一项所述的另一些实施方案中,所述ago蛋白包括核定位信号(nls)。
实施方案110:在一些实施方案中,提供了一种细胞,所述细胞包含根据实施方案75、79和87-109中任一项所述的组合物。
实施方案111:在一些实施方案中,提供了一种试剂盒,所述试剂盒包含根据实施方案75、79和87-109中任一项所述的组合物以及使用所述试剂盒修饰靶核酸的说明书,其中所述靶核酸包含所述靶基因座。
实施例
以下实施例旨在仅对本发明进行举例说明并且因此不应被视为以任何方式限制本发明。
实施例1:ago-gdna复合物对靶质粒的体外切割
在该实施例中,将各种菌种的argonaute蛋白分离并亚克隆到表达质粒中,使该argonaute蛋白与引导dna一起共转染到293t细胞中,以提供gdna预负载的ago复合物(“ago-gdna”)。将纯化的ago-gdna复合物各自在体外与靶质粒一起温育,以在不同温度下评估ago的dna-引导的dna内切核酸酶活性。
1.ngago
通过pcr使用引物flag-hindiii-f和ha-bamhi-r,从嗜盐嗜碱古细菌格氏嗜盐碱杆菌sp2的基因组dna分离ngago的完整编码序列,用限制性酶hindiii和bamhi切割,并亚克隆到pcdna3.1/hygro(+)质粒(invitrogen)中,以提供质粒flag-ngago-ha-pcdna3.1。flag和ha标签序列分别融合于ngago的编码序列的5′和3′。ngago的氨基酸序列(seqidno:1)如下所示:
seqidno:1(ngago)
5′磷酸化单链引导dna(“fw引导物”,表2)被设计成与pacycduet-egfp质粒(novagen)中gfp基因的靶位点互补(图1)。
将flag-ngago-ha-pcdna3.1质粒和fw引导物共转染到人293t细胞中。使用flag、ha串联亲和纯化试剂盒(sigma-aldrich),从共转染的细胞纯化包含结合引导dna的flag-ngago-ha蛋白质的复合物(“ngago-gdna”)。简而言之,在flag-ngago-ha-pcdna3.1质粒和引导dna转染后48小时,将5×107个293t细胞用5mlripa缓冲液裂解并在冰上超声处理。在离心后,收集上清液并与100μl抗-flagm2树脂混合,然后在4℃下轻柔摇动过夜。然后,将树脂用1mlripa洗涤一次,并用1ml不含bsa的反应缓冲液(体外裂解测定)洗涤3次。用300μl0.2mg/ml3×flag肽(溶于不含bsa的反应缓冲液中)从树脂洗脱flag-ngago-ha。通过page检查洗脱物来评估质量和量。如果flag-ngago-ha蛋白质高于80%纯度,则进行超滤以使flag-ngago-ha蛋白质浓缩至约1mg/ml以供随后酶分析;如果样品的纯度不理想,则使用100μl抗-ha树脂重复如上所述的纯化步骤。
在4℃、20℃、30℃、40℃、50℃、或60℃下,将5μg的经纯化ngago-gdna复合物与400ngpacycduet-egfp质粒在50μl反应混合物(10mmtrisph8.0,20mmnacl,0.5mmmgcl2,0.4%甘油,2mmdtt和20mg/μlbsa)中一起温育8小时。然后,将反应混合物在52℃下用蛋白酶k处理2小时,以降解反应混合物中的蛋白质,并在1%琼脂糖凝胶中通过电泳分析所得产物。
ngago-gdna复合物的体外质粒切割测定结果示出于图2a和图2b。超螺旋(sc)态(即未裂解)、线性化(lin)态(即两条链被切割)和开环(oc)态(即一条链被切割)的质粒的相对尺寸示于“m1”标记的泳道中,并且分子量标记示于泳道“m2”中。如图2a所示,在与ngago-gdna复合物温育4小时后,仍然存在未切割的超螺旋和单链切割的开环物类,而一些质粒两条链被切割而产生线性化形式。在8小时和72小时之后,质粒仅显示为线性化dna,这表明ngago能够使用单一预负载的引导dna完全切割质粒的两条链。类似地,ngago-gdna复合物能够诱导靶质粒的双链断裂,以在除4℃之外的所有测试温度下得到线性化产物(图2b)。
为了确定fwgdna预负载的ngago体外裂解之后质粒上切割位点的位置,在温育72小时后从琼脂糖凝胶切下线性化质粒,并通过凝胶提取纯化。在纯化dna的pcr聚腺苷酸化之后,将所得产物克隆到pgem-t-载体(promega)中。氯霉素和氨苄青霉素双抗性克隆被选择用于质粒提取,然后测序(sangonbiotech)。
图2c在顶部示出pacycduet-egfp质粒的野生型靶位点的序列,与下面的温育72小时之后ngago-gdna的质粒切割产物的四个代表性测序结果进行比对,并且fw引导物序列加阴影。还示出了两个代表性的测序色谱图,并且序列对应于以下所示的pgem-t载体、fw引导物和pacycduet-egfpdna。序列是20个独立实验的代表性结果。结果示出靶位点被ngago成功地切割。除简单地断裂单个磷酸二酯键之外,ngago还随机地移除靶位点内的1至20个核苷酸。靶位点处核苷酸的移除不由于ngago的外切核酸酶活性。如图2b所示,即使在相同条件下8小时后实现完全线性化,使酶促反应延长至72小时不导致线性化靶标的进一步降解。该发现符合ttago反应,其也移除了其靶标中的核苷酸(wang,y.等人nature461,754-761(2009))。
为了进一步研究ngago是否能够切割线性化dna,通过采用bamhi限制性酶在位于质粒的限制性位点处进行切割,使pacycduet-egfp质粒线性化(图3a,顶部)。在37℃下,使线性化质粒与或不与从293t细胞纯化并预负载fw的ngago(“ngago-fw-g”)共温育8小时。当线性化质粒与或不与ngago-fw一起温育时,不存在可识别的切割(图3a,分别为泳道2和1)。
还测试了ngago-fw-g切割86个核苷酸ssdna片段(seqidno:167)的能力。在37℃下,将86ntssdna与或不与ngago-fw-g共温育8小时。结果表明,不存在86nt靶序列的切割(图3b,分别为泳道2和1)。泳道m在左侧显示具有以nt计所示尺寸的分子量标记。这些数据表明,ngago不是外切核酸酶。
2.naago
将亚洲嗜盐碱杆菌argonaute(“naago”)基因序列从细菌亚洲嗜盐碱杆菌dsm12278(atccatcc保藏号700177;cgmcc(中国普通微生物菌种保藏管理中心,chinesegeneralmicrobiologicalculturecollectioncenter)保藏号1.2199)的基因组dna分离,并进行亚克隆,以得到如上所述的flag-naago-ha-pcdna3.1质粒。naago的氨基酸序列(seqidno:4;ncbi登录号wp_006111085.1)示于下文。
seqidno:4(naago)
将flag-naago-ha-pcdna3.1和fw引导dna共转染到293t细胞中,并将naago-gdna复合物纯化并与如上所述的靶pacycduet-egfp质粒一起温育。体外切割反应的电泳示出,naago-gdna复合物能够诱导靶质粒的双链断裂,以在除4℃之外的所有测试温度下产生线性化产物(图4a)。
3.ntago
将西藏嗜盐碱红菌argonaute(“ntago”)基因序列从细菌西藏嗜盐碱红菌ga33(cgmcc登录号1.2123)的基因组dna分离,并进行亚克隆,以得到如上所述的flag-ntago-ha-pcdna3.1质粒。ntago的氨基酸序列(seqidno:5;ncbi登录号wp_006090832.1)示于下文。
seqidno:5(ntago)
将flag-ntago-ha-pcdna3.1和fw引导dna共转染到293t细胞中,并将ntago-gdna复合物纯化并与如上所述的靶pacycduet-egfp质粒一起温育。体外切割反应的电泳示出,ntago-gdna复合物能够诱导靶质粒的双链断裂,以在除4℃之外的所有测试温度下产生线性化产物(图4b)。
4.maago
将铜绿微囊藻argonaute(“maago”)基因序列从细菌铜绿微囊藻nies843(fachb登录号315)的基因组dna分离,并进行亚克隆,以得到如上所述的flag-maago-ha-pcdna3.1质粒。maago的氨基酸序列(seqidno:2;ncbi登录号wp_012265209.1)示于下文。
seqidno:2(maago)
将flag-maago-ha-pcdna3.1和fw引导dna共转染到293t细胞中,并将maago-gdna复合物纯化并与如上所述的靶pacycduet-egfp质粒一起温育。体外切割反应的电泳示出,ntago-gdna复合物能够诱导靶质粒的双链断裂,以在除4℃之外的所有测试温度下产生线性化产物(图4c)。
5.syago
将聚球藻argonaute(“syago”)基因序列从细菌聚球藻7942(fachb登录号805)的基因组dna分离,并进行亚克隆,以得到如上所述的flag-syago-ha-pcdna3.1质粒。syago的氨基酸序列(seqidno:12;ncbi登录号wp_011378069.1)示于下文。
seqidno:12(syago)
将flag-syago-ha-pcdna3.1和fw引导dna共转染到293t细胞中,并将syago-gdna复合物纯化并与如上所述的靶pacycduet-egfp质粒一起温育。体外切割反应的电泳示出,ntago-gdna复合物能够诱导靶质粒的双链断裂,以在除4℃之外的所有测试温度下产生线性化产物(图4d)。
实施例2:大肠杆菌中表达的ngago对靶质粒的体外切割
在该实施例中,从大肠杆菌中表达并纯化ngago,并与引导核酸一起温育,以评估其体外内切核酸酶活性和需要。
gst-ngago在大肠杆菌中的表达和纯化
为确认ngago利用ssdna引导物,使ngago表达于大肠杆菌中,因为这些细菌产生5′磷酸化ssrna和ssdna两者。gst-标记的重组ngago蛋白如下产生:使用引物ngago-6p-1f和ngago-6p-1r,使用pcr从格氏嗜盐碱杆菌sp2的基因组dna分离ngago,用限制性酶bamhi、bg1ii和xhoi切割,并克隆到pgex6p-1质粒(gehealthcare)中。使ngago-6p-1质粒(图5)转化到大肠杆菌菌株bl21de3(clontech)中以表达gst-ngago蛋白。用还原的谷胱甘肽缓冲液洗脱琼脂糖凝胶-4b-纯化的gst-ngago。
结合细菌衍生的gst-ngago的核酸的电泳
将纯化的蛋白质在52℃下用蛋白酶k消化2h。通过roti酚、氯仿、异戊醇(ph7.5-8.0,carlrothgmbh)从蛋白质分离核酸并利用乙醇沉淀进一步纯化。使来自5ml蛋白溶液的空气干燥的沉淀物重悬于100μl蒸馏水中,然后经受dna酶i或rna酶a消化。然后将核酸用roti酚、氯仿、异戊醇纯化并用异丙醇沉淀。在重悬于50μlte缓冲液(ph8.0)之后,通过变性page分析核酸并利用银染可视化。
结合gst-ngago的核酸的银染示出,样品中存在的大小为约20-30个核苷酸的条带没有经受消化(图6,从左侧的第二泳道)。采用rna酶a进行核酸酶消化不导致这些条带消失(图6,左侧的第三泳道)。当经受dna酶i时,核酸被消化(图6,右边两个泳道)。左泳道为单链分子量标记(m),显示核苷酸的大小(nt)。这证实ngago结合了ssdna。
各种引导dna的体外切割测定
为测试大肠杆菌表达的ngago是否可在ssdna引导物存在时切割pacycduet-egfp质粒的靶位点(图1),进行体外质粒切割测定。在切割测定之前,将结合纯化gst-ngago的天然核酸用设计的ssdna引导物(fw、rv、或nc,图1)替代。pacycduet-egfp质粒(novagen)不具有gst-ngago-编码质粒pgex6p-1的同源序列。各个ssdna引导物是5′磷酸化的,并且长度为24个核苷酸(nt)。两种引导物即fw和rv彼此互补并与靶位点序列互补。非互补引导物(nc)包含不与pacycduet-egfp序列重叠的随机序列。
为了重负载引导物,在55℃下,使5μggst-ngago与300ng核酸引导物在50μl反应缓冲液(10mmtrisph8.0,20mmnacl,0.5mmmgcl2,0.4%甘油,2mmdtt和20μg/mlbsa)中一起温育1h。使400ngpacycduet-egfp质粒溶解于20μl反应缓冲液中,并添加到ngago溶液中在37℃下温育8h。随后,将混合物用蛋白酶k在52℃下处理2h以移除蛋白质,然后在1%琼脂糖凝胶中经受电泳。示出了超螺旋(sc)、线性化(lin)和开环(oc)质粒的相对尺寸(图7a,左泳道,“m1”)。
当单独或有非互补nc引导ssdna(图7a,分别为泳道2和3)存在条件下,ngago不切割负超螺旋质粒dna。当供应有互补fw或rv引导物时,ngago使负超螺旋质粒dna的一条链断口,从而允许围绕磷酸二酯主链旋转并产生开环构象(图7a,分别为泳道5和6)。当供应有fw和rv引导物两者时,ngago切割质粒dna的两条链,使质粒线性化(图7a,泳道7)。不同于引导物重负载的ngago,负载一种引导dna的ngago可在体内有效地线性化靶质粒,表明ngago在37℃下负载单一引导物时使得双链断裂,但在55℃下重负载引导物时并非如此。不受理论或假设的约束,ngago在55℃下处理时变得部分变性并丧失一些活性。实际上,将大肠杆菌与引导dna在55℃下延长温育72小时导致其核酸酶活性完全丧失(图7b)。
在未5′磷酸化的ssrna引导物和ssdna引导物存在情况下进一步测试ngago的体外切割活性。当供应有或没有5′磷酸化的ssrna时,ngago不能切割质粒(图7c,分别为泳道3和2)。当供应未5′磷酸化的ssdna(图7c,泳道4)时,其也不能切割质粒。仅当ngago供应有5′磷酸化ssdna引导物时,超螺旋质粒线性化。因此,仅当供应有5′磷酸化ssdna引导物时,ngago能够在37℃下切割双链dna靶标。
实施例3:人293t细胞中ngago的体内引导物负载
该实施例评估了在体外和293t细胞中ngago的引导核酸负载。
ngago和引导核酸的同时转染
将flag-ngago-ha-pcdna3.1质粒与或不与各种引导核酸同时转染到人293t细胞中。然后如实施例1中所述,从293t细胞纯化flag-ngago-ha蛋白。分离相关核酸并通过电泳进行分析。
如图8a泳道1中所示,在外源核酸没有共转染的情况下,不存在可检测的与从293t细胞纯化的ngago相关联的内源核酸。这意味着在人细胞中存在非常有限的5′磷酸化ssdna,表明内源性ssdna不误导ngago从位点脱靶。
为确定ngago是否可与人细胞中的外源核酸关联,将293t细胞用ngago编码质粒与有或没有5′磷酸化的合成24-ntssdna或ssrna寡核苷酸共转染。ngago仅可与5′磷酸化ssdna(图8a,泳道5)相关联,但未5′磷酸化的ssrna(泳道2和3)或ssdna(泳道4)并非如此。泳道m1示出ssdna梯度并且m2是ssdna引导物的阳性对照。
ngago之后用引导dna转染
为了进一步检查ngago与5′-磷酸化ssdna在人细胞中的缔合时序,首先将flag-ngago-ha-pcdna3.1转染到293t中,并在编码ngago的质粒转染之后,使5′-磷酸化ssdna(fw引导物)向细胞的递送延迟0h、12h或24h(分别为图8b,泳道2、3和4)。当fw引导物在flag-ngago-ha-pcdna3.1转染后12h转染到293t细胞中时,fw引导物结合ngago的能力并未显著下降(图8b,泳道3)。然而,当fw引导物在延迟24h后转染时,ngago-ssdna结合显著降低,这表明当表达ngago时ssdna负载发生于较短的时间窗口内(图8b,泳道4)。m1示出ssdna梯度,m2示出阳性对照fwssdna,并且泳道1示出ngago在无fw引导dna下温育。
纯化ngago与引导dna的共温育
相比之下,ngago仅在升高的温度下在体外有效负载引导dna。使flag-ngago-ha-pcdna3.1质粒转染到细胞中而无需任何外源核酸。然后从293t细胞纯化flag-ngago-ha,并且在55℃下与fwssdna一起温育1h或在37℃下温育8h。如图8c(泳道3)所示,即使在温育8小时之后,ssdna也不能在37℃的生理温度下于体外负载到293t-纯化的ngago上。在55℃的非生理温度下,fw引导物在1小时后负载于ngago上(图8c,泳道2)。图8c泳道m1示出了ssdna梯度(左侧示出核苷酸的大小(nt)),泳道m2示出阳性对照fwssdna,并且泳道1示出ngago在无fw引导dna下温育。
ngago-gdna复合物的体外切割活性
如实施例1中所述,在体外切割测定中对体内或体外引导物负载所获得的各种ngago-gdna复合物进行测试。源于与靶互补fw引导物(图8d,泳道4)共转染的细胞,而非源于与随机nc引导物(图8d,泳道5)共转染的细胞的ngago,可导致双链断裂并使质粒线性化。即使纯化的ngago随后与fw引导物共温育,源于无引导物共转染的细胞的ngago也不可切割靶标(图8d,泳道3)。此外,即使是随后在37℃下与fw引导物一起在体外温育8小时,来源于具有非特异性ssdna引导物(诸如nc)的细胞的ngago也不可切割质粒dna(图8d,泳道6)。这些数据表明,ngago忠于其初始引导物,并且不允许在37℃下进行gdna交换。该种ngago特性使其将负载意料不到的“引导物”的可能性最小化。类似地,哺乳动物ago2蛋白不可在37℃用游离寡核苷酸交换其结合的寡核苷酸(elkayam,e.等人cell150,100-110(2012);schirle,n.t.,sheu-gruttadauria,j.¯ae,i.j.science346,608-613(2014))。
ngago所遵循的“一种-引导物-忠实性”规则
图9汇总了基于上述引导物负载实验的“一种-引导物-忠实性”特性。图9的顶部图示出ngago表达质粒转染到其表达产生ngago蛋白的哺乳动物细胞中。在从细胞纯化ngago蛋白之后,将其与fw引导物ssdna共温育,但这不导致活性质粒切割能力。这说明ngago不可在37℃下负载ssdna引导物(图8c,泳道3),并且纯化ngago与fw引导物共温育不导致质粒切割活性(图8d,泳道3)。
图9的中间图示出ngago表达质粒与5′磷酸化和未磷酸化fwssdna和ssrna引导物共转染到哺乳动物细胞中。这些细胞中表达的ngago蛋白仅可结合5′磷酸化fwssdna引导物,并且当从细胞纯化ngago-gdna复合物时,其在质粒切割中呈活性。这示出了5′磷酸化fwssdna引导物与纯化ngago缔合(图8a,泳道5),但不与未磷酸化fw(图8a,泳道4)或ssrna(图8a,泳道2和3)缔合。该示意图还示出体内预负载的ngago-fwgdna复合物在体外切割质粒dna的能力(图8d,泳道4)。
图9的底部图示出ngago表达质粒与非互补ncssdna引导物共转染到哺乳动物细胞中。ngago蛋白与nc引导物缔合,但即使是与fw引导物在体外一起温育之后,纯化的ngago-ncgdna随后也不能切割质粒dna。这示出了图8d泳道5和6中所示的结果。
实施例4:ago-gdna对靶质粒的细胞内修饰
该实施例描述了ago-gdna在hela细胞中对靶质粒的细胞内活性。
具有核定位的ngago的构建体
为测试ngago修饰人细胞中靶核酸的能力,修饰的ngago通过使核定位信号(nls)连接于其n-末端来设计,以确保核区室化。将nls-ngago构建体克隆到pcdna3.1质粒中以提供nls-ngago-pcdna3.1质粒。
为可视化nls-ngago蛋白的定位,产生荧光蛋白dsred融合于nls-ngago构建体的nls-ngago-red质粒。使用引物nls-ngago-hindiii-f2和nls-ngago-bamhi-r,利用pcr从格氏嗜盐碱杆菌sp2的基因组dna分离ngago,用限制性酶hindiii和bamhi切割,并亚克隆到pdsred-monomer-n1质粒(clontech)中(表4)。将工程化nls-ngago转染到hela细胞中。使用明视场显微镜图像使细胞可视化(图10,右侧),利用dapi染色使核可视化(中间),并通过荧光显微法检测nls-ngago-red。结果表明,nls-ngago位于hela细胞的细胞核中。
ngago-gdna和cas9-sgrna对靶质粒的活性的比较
为研究ngago-gdna系统是否是哺乳动物细胞中定向dna修饰的有效工具,进行细胞内质粒切割测定。ssdna引导物被设计成用于靶向pegfp-n1质粒上的两个区域。图11a示出了环化pegfp-n1质粒的示意图,ssdna引导物g1、g2的定位在cmv启动子编码区之内,并且ssdna引导物g3和g4定位在egfp基因之内。为了比较ngago-gdna系统与cas9-sgrna系统,还设计了cas9靶向pegfp-n1质粒的单一引导sgrna。表2示出了5′磷酸化ngagossdna引导物g1、g2、g3和g4、阴性对照非互补引导ncgssdna的序列,以及编码cas9单一引导sgrna序列sgrna1和sgrna2的dna序列。图11b示出线性化pegfp-n1质粒的示意图,ngago引导物和cas9引导物的位置相对于靶cmv启动子和egfp基因示出。
为了在哺乳动物细胞中比较ngago-gdna系统与cas9-sgrna系统的靶质粒修饰效率,使两种系统转染到hela人上皮细胞系中。简而言之,在24孔板中使用
当细胞用仅ngago、仅g3引导物、或ngago和非互补ncg引导物两者共转染时,由hela细胞中靶pegfp-n1质粒表达的egfp蛋白质的量不降低。(图11c,泳道2-4)然而,当细胞用pegfp-n1、ngago、以及任何ssdna引导物(g1、g2、g3和g4)共转染时,egfp的表达水平显著下降(图11c,泳道5-8)。相比之下,当相比于用单独的靶pegfp-n1和spcas9转染的细胞时(图11c,泳道11),用spcas9编码质粒以及编码sgrna1(cmv)或sgrna2(egfp)的sgrna转录载体转染的细胞中egfp的表达水平仅适度下降(图11c,泳道9-10)。因此,当提供具有相同量ngago和cas9质粒的细胞时,ngago-gdna系统如同cas9-sgrna系统有效地抑制egfp表达。
细胞内活性的最佳gdna长度
为确定ssdna引导物在引导ngago进行质粒修饰方面的最佳长度,使引导dna(g3(n)引导物)基于g3引导物进行设计,但长度从20个核苷酸到27个核苷酸变化。将hela细胞用pegfp-n1质粒、nls-ngago-pcdna3.1质粒以及各g3(n)引导物进行转染。在转染后36小时,收获细胞,并通过蛋白质印记杂交分析细胞溶解物以测定egfp的表达水平。尽管效率有所不同,具有20-27个核苷酸的gdna可全部结合ngago,并允许切割靶dna(图11d)。用24-nt引导dna转染的细胞示出egfp表达的最大抑制,这表明24个核苷酸是ngago的引导dna的最佳长度(图11d,泳道6)。
其它ago-gdna系统对靶质粒的修饰
使用类似于如上所述ngago的方法,maago、hpago、naago、ntago、npago、hbago、miago、syago、lyago、scago、teago、phago、mlago、hmago、bgago、baago和rbago被发现在hela细胞中对靶质粒pegfp-n1有类似的修饰效率(图12)。在这些实验中使用g3gdna。相比之下,ttago(末图)不能抑制hela细胞中的gfp表达,因为该酶获自嗜热细菌菌种并且仅升高的温度呈活性。hpago、npago、hbago、miago、lyago、scago、teago、phago、mlago、hmago、bgago、baago和rbago的氨基酸序列示于下文。
seqidno:3(hpago)
seqidno:6(npago)
seqidno:7(hbago)
seqidno:11(miago)
seqidno:33(lyago)
seqidno:28(scago)
seqidno:9(teago)
seqidno:41(phago)
seqidno:40(mlago)
seqidno:39(hmago)
seqidno:38(bgago)
seqidno:37(baago)
seqidno:42(rbago)
实施例5:哺乳动物细胞中ngago-gdna的内源性基因座切割
该实施例描述ngago-gdna切割人基因组中内源性位点的能力。使用如实施例7中所述的t7ei测定,通过非同源末端连接(nhej)途径,基于由双键断裂的缺陷修复(即插入缺失突变)引起的突变率,对ngago-gdna的切割活性进行定量。将ngago-gdna的基因组切割效率与cas9-sgrna的基因组切割效率进行比较。
293t细胞中dyrk1a基因座的切割
五种gdna被设计成靶向人dyrk1a基因的外显子11。图13a示出了ssdna引导物g5、g6、g10、g12和g13的序列(表2),以及它们沿人dyrk1a基因座的序列的靶向位置。将293t细胞用nls-ngago-pcdna3.1质粒和指定gdna共转染。提取基因组dna,并使用引物dyrk1a测试f和dyrk1a测试r(表3)通过pcr进行扩增,得到584bp产物。对于各个t7ei反应,使500ng的pcr产物变性,再退火并用t7内切核酸酶i消化,该酶切割由于ngago-gdna诱导的dsb的nejh修复的错配异源双链核酸分子。通过变性page分析反应并利用银染可视化。
图13b示出了显示人dyrk1a基因的ngago-gdna切割的t7ei测定结果。对照(泳道1)示出来自293t细胞基因组dna的未修饰584bppcr产物,并且分子量标记在泳道m中示出。所有gdna均能够引导ngago高效靶切割(泳道3-7)。箭头显示t7ei切割产物的位置。ngago-gdna在dyrk1a基因座处的切割效率介于27.3%和39.1%之间。
使用g10引导物对克隆扩增子的突变等位基因的测序确认,插入缺失在ngago-gdna切割之后引入靶基因座。图13c示出突变等位基因的代表性序列(底部图)与具有10bp微缺失(由省略号标记的位置)的实施例色谱图(顶部图),该实施例色谱图对应于与wtdyrk1a序列比对的序列d10。
为了进一步验证t7ei测定,不同的引物对用于dyk1a基因座上的t7ei测定。图14a示出注释有引物退火位点、g5和g10引导dna、以及sg-drk1asgrna和对应的t7ei切割位点的dyrk1a基因座的示意图。图14b示出展示ngago-gdna或cas9-sgrna系统切割的t7ei测定结果。t7ei切割产物具有预测的长度。
293t细胞中其它基因座的切割
另外使用靶向8种哺乳动物基因组基因座(actin、emx1、hba2、gata4、grin2b、hres1和apoe)的47种引导dna来测试ngago-gdna系统。各基因座的切割效率使用t7ei测定法来确定。引导dna的序列在表2中列出。扩增靶区域的引物和扩增产物的长度的引物列于表3中。
图15a中的t7ei测定结果展示ngago-gdna对dyrk1a、emx1、grin2b、gata4和hba2的切割。顶部条带表示未切割的dna,并且箭头指示t7ei切割产物。对于所有测试的基因座,切割效率类似(24.5%-30.1%)。本实验中使用的引导dna分别为g10(dyrk1a)、g31(emx1)、g43(grin2b)、g40(gata4)和g37(hba2)。
图15b中的t7ei测定结果展示ngago-gdna对hba2、gata4、grin2b、hres1和apoe的切割。对于测试的所有引导dna和所有基因座,切割效率相当(21.3%-41.3%)。
对于使用靶向dyrk1a(图15c)、actin(图15d)和emx1(图15e)的各种ssdna引导物的ngago-gdna系统,对t7ei测定所测得的插入缺失百分比进行进一步比较。切割效率对于所有引导dna是相当的,并且没有观察到ngago对具有特定性质的序列有偏好。
不同细胞系中dyrk1a基因座的切割
在不同的人细胞系中测试ngago-gdna对dyrk1a基因座的切割效率。用nls-ngago-pcdna3.1质粒和ssdna引导物g10转染各细胞系。进行t7ei内切核酸酶测定来测定切割效率。如图16所示,ngago-gdna系统可有效切割293t(人胚肾)、mcf7(人乳腺癌)、k562(人骨髓)和hela(人上皮)细胞中的dyrk1a基因座。
使用错配gdna切割dyrk1a基因座
为研究gdna与靶序列之间的核苷酸错配对ngago活性的影响,将单个核苷酸和三个核苷酸错配引入靶向dyrk1a基因座的24-ntgdnag10。图17a示出24-ntg10ssdna引导物序列(顶部)与各自被设计成包含单个错配核苷酸的24种不同的引导物(m1-m24,错配的核苷酸以下划线标注),以及各自具有连续三个核苷酸错配的3种引导dna(m25-m27)的比对。t7ei测定结果示出,ngago-介导的靶切割对g10引导物的各位置处的单个核苷酸错配较敏感。当ngago-g10gdna导致30.4%插入缺失(g10泳道,最右侧)时,使用错配的引导物使切割效率降至约0至8.1%。这反映出73-100%的切割效率降低,使用引导物m8-m11观察到最大程度的降低。此外,任何测试位点处三个连续核苷酸的错配完全破坏切割。
另外,基于靶向相同的dyrk1a基因座的缩短的21-nt长ssdna引导物g10来设计各种错配的gdna(m1-m24),以测试核苷酸错配对ngago介导的靶切割的效率的影响。图17b中示出t7ei测定的结果。类似于24nt长gdna的结果,切割效率对gdna与靶基因座之间的单个核苷酸错配较敏感,并使用gdnam8-m11观察到效率的最大程度降低。使用具有连续三个核苷酸错配(m22-m24)的gdna观察到较小切割。
ngago-gdna与cas9-sgrna之间切割效率的比较
ngago-gdna在靶向富gc基因座中优于cas9-sgrna,这是由于其依赖于dna引导物而非rna引导物。rna(而非dna)易于形成二级结构,其可改变构象并阻断结合靶基因座。为了比较ngago-gdna和cas9-sgrna在切割富gc区中的效率,在hba2和gata4基因座上测试这些系统。
hba2基因包含跨越361-600位核苷酸(70.8%富gc)的基因座。图18a顶部图示出具有加下划线的靶位点的hba2基因座的序列,下方对准的对应sgrna(hba2)序列、以及g37gdna序列。gata4基因包含跨越核苷酸31441-31680位(78.8%gc)的基因座。图18a底部图示出了具有加下划线的靶位点的gata4基因座的序列,上方对准的有对应sgrna(gata4)序列、以及g40gdna序列。进行t7ei测定来测定各个基因座处的切割效率。
图18b中示出t7ei测定的结果。使用未切割的293tdna作为对照(“ck”泳道),并且切割产物用箭头指示,其中插入缺失百分比在泳道下方示出。spcas9-sgrna导致不可检测到的hba2基因座的切割(左侧的泳道4)。ngago-gdna以37.5%的效率切割相同的富gc基因座(左侧的泳道3)。cas9-sgrna能够以13.1%的效率切割gata4基因座,而ngago-gdna以31.5%的更高效率切割相同的基因座(最左侧两个泳道)。因此,与在富gc基因座处具有受限切割活性的cas9-sgrna系统相比,ngago-gdna系统更有效地切割富gc基因座。另外,而且cas9要求靶基因座紧靠着pam序列的上游存在,argonaute基于我们的观察和其它报道(swarts,d.c.等人nature507,258-261(2014);swarts,d.c.等人nucleicacidsres.43,5120-5129(2015))不具有靶序列限制。因此,相比于cas9-sgrna系统,ngago-gdna可应用于更广泛的基因组基因座。
实施例6:ngago-gdna介导的哺乳动物细胞中的基因组编辑
在真核细胞中,双链断裂(dsb)主要通过两种途径修复:非同源末端连接(nhej)和同源定向重组(hdr)。因为ngago-gdna可诱导哺乳动物细胞基因组dna的双链断裂,可利用内源性nhej或hdr修复途径使用ngago-gdna作为基因组编辑工具。
hdr介导的基因组编辑
hdr是一种用于敲入、或捕获供体dna以产生基因组的特异性修饰的广泛使用的策略。为测试ngago-gdna系统是否可用于引发hdr介导的基因组编辑,将供体dna片段设计成靶向人dyrk1a基因的外显子11。
图19a示出通过ngago-gdna进行hdr介导的供体dna插入的示意图。mrfp-tga-egfp供体dna被设计成包含报告基因区域(mrfp-tga-egfp),其具有两个阅读框,即由tga终止子隔开的mrfp和框外egfp基因。报告基因区域无启动子。供体dna还包含受cmv启动子(cmv-g418r-pa)控制的g418抗性基因以利于编辑细胞的选择。报告基因侧接与dyrk1a基因的外显子11同源的5′臂和3′臂。注释的供体dna的序列示出图19a的顶部。不受任何假设理论的约束,ngago-gdna可在dyrk1a中诱导dsb,引发同源介导的修复(hdr),其经由同源臂在dsb处捕获供体dna。这导致供体序列插入dyrk1a基因的外显子11中。在供体dna框内插入靶基因座中时,报告基因区域由dyrk1a启动子驱动,因此mrfp和g418被表达。然而,因为tga终止子,egfp不被表达。
为了整合mrfp-tga-egfp构建体,采用200ngnls-ngago-pcdna3.1质粒、100ngg10ssdna引导物和500ngmrfp-tga-egfp供体dna,使用
测序结果确认在ngago-gdna切割之后,供体dna准确插入到dyrk1a基因座中。5′接头的示例性测序色谱图示出包含5′供体臂和mrfp-tga-egfp供体的起始部的序列(图19b顶部)。3′接头的示例性测序色谱图示出包含供体dna的端部和3′供体臂的序列(图19b,底部)。
随后用g418对细胞进行处理以进行阳性克隆选择。在g418处理的两周之后,大多数细胞表达mrfp。分离具有mrfp表达的细胞菌落。
nhej介导的基因组编辑
接着使具有稳定整合的mrfp-tga-egfp报告基因的阳性克隆经受靶向tga终止子位点的第二ngago-gdna复合物,以展示nhej介导的基因组编辑。此处,整合的报告基因区域(图19c)包含驱动mrfp基因表达的启动子,继之以g52靶序列、tga终止密码子和框外egfp基因,使得细胞克隆仅表达mrfp。ngago-g52gdna复合物可在tga终止密码子的g52靶位点上游处诱导双链断裂。不受任何理论或假设的约束,可通过nhej在细胞内不精确地修复dsb,并且在一些情况下,可在靶位点处引入移码突变,从而使终止密码子移码并使egfp移入框中。因此,可产生表达mrfp和egfp两者的细胞。
使nls-ngago-pcdna3.1质粒和g52ssdna引导物共转染到稳定的mrfp-tga-egfp整合的细胞中。两天后,收获细胞并进行流式细胞术分析,以评价mrfp-egfp融合蛋白的表达。用空载体、单独的nls-ngago-pcdna3.1、或单独的g52gdna转染的细胞仅显示mrfp表达(图19d)。然而,当mrfp-tga-egfp细胞用nls-ngago-pcdna3.1质粒和g52gdna转染时,11.7%的细胞表达rfp和gfp两者(图19d,右侧),表明框外突变成功引入g52靶位点。
ngago-gdna和cas9-sgrna的脱靶基因组编辑
为研究ngago-gdna和cas9-sgrna的脱靶效应,从egfp基因扩增400bp的dna片段,以产生与dyrk1a靶标没有任何同源性的egfp400供体dna。使用引物egfp79和egfp483从degfp-n1质粒pcr扩增egfp400供体dna(表3)。将293t细胞用ngago-gdna系统(nls-ngago-pcdna3.1质粒和g10引导物)或cas9-sgrna系统(spcas9-pcdna3.1和sgrna(dyrk1a)载体)与egfp400供体dna一起转染。图20a(顶部图)示出与g10gdna和sgrna对准的人dyrk1a基因的外显子11中的基因座的序列。ngago-gdna和cas9-sgrna系统可在靶基因座处诱导dsb,并且egfp400供体dna可整合进基因组的dyrk1a靶位点内(命中靶)或其它地方(脱靶)(图20a,底部)。
从转染的细胞提取总基因组dna,使用限制性酶消化,并通过southern印迹杂交分析进行分析。简而言之,使用引物egfp79和egfp483利用pcr产生探针。然后使用克列诺片段(newenglandbiolabs),采用32p-dntp标记pcr产物。在转染后48小时,收集工程化293t细胞,并根据制造商的说明书使用dneasy试剂盒(qiagen)分离总细胞dna。将约2.5μgdna用dna核酸内切酶bglii、sali、saci、xhoi、af1ii和eco47iii(newenglandbiolabs)消化。通过0.6%琼脂糖凝胶分离消化的片段。然后将dna转移至尼龙膜并进行uv交联。在与鲑精dna(约100个碱基对)预杂交之后,将dna与32p-标记的探针在55℃的杂交缓冲液(7%sds,0.5m磷酸钠缓冲液,ph7.4)中一起温育8h。然后将膜用含有0.1%sds的1×ssc洗涤三次,并暴露于x射线胶片。
图20b示出southern印迹杂交。32p-标记的探针不与对照样品(泳道1)、或仅经ngago编码质粒转染的细胞的dna(泳道2)杂交。泳道6示出与gfp-n1阳性对照杂交的探针。bg1ii反应产生包含中靶片段的6.5kb片段,其在经ngago和g10gdna、或cas9和sgrna转染的细胞的dna中检测出(泳道4和5)。在表达cas9的细胞中检测到对应于脱靶整合的高分子量条带(泳道3,5),但在表达ngago的细胞中未检测到。
内源性基因座中敲入egfp供体dna
通过使用ngago-g10gdna、phago-g10gdna、miago-g10gdna、或maago-g10gdna将egfp供体dna插入人dyrk1a基因的外显子11的靶基因座(seqidno:153,154)中,实施第二敲入实验。另外,使用ngago-g21gdna或phago-g21gdna,将egfp供体dna敲入人β-肌动蛋白基因的不同靶基因座中。
使用phago-g10gdna作为示例,图21a示出ngago-gdna介导的egfp供体dna在dyrk1a基因座处的示意性敲入。编码ngago蛋白的质粒经密码子优化以在人细胞中表达,并且ngago的n-末端和c-末端各自融合于nls拷贝。egfp供体dna被设计成包含无启动子egfp编码基因、以及sv40polya信号,其侧接与dyrk1a基因的外显子11同源的5′臂和3′臂。使用供体f和供体r引物和seqidno:155的egfp模板扩增egfp供体dna。不受任何理论或假设的约束,ngago-gdna在dyrk1a中诱导dsb,引发同源介导的修复(hdr),并经由其同源区插入供体dna。为敲入到β-肌动蛋白基因座中,使用β-肌动蛋白测试f引物(seqidno:114)和β-肌动蛋白测试r引物(seqidno:115)和seqidno:155的egfp模板扩增egfp供体dna。
为了整合egfp供体dna,将24孔板各个孔中的293t细胞在第一转染中用脂质转染胺与300ngnls-ago质粒、300ng引导dna进行转染,并在第一转染后20小时,在第二转染中使用300ngegfp供体dna进行转染。在第二转染后48-60小时,从细胞提取基因组dna。用引物dy1和g1r对修饰的dyrk1a基因组基因座进行pcr扩增,并且采用引物肌动蛋白测试f和g1r对修饰的β-肌动蛋白基因组基因座进行pcr扩增(表3)。对于每50μl的pcr反应,将1μl的基因组dna(200ng/μl)、1.5μl的正向引物(10μmol/μl)、1.5μl反向引物(10μmol/μl)、以及25μl的2xtaqpcrstarmix(genstar)和21μlh2o混合。使用引物dy2和g2r(用于修饰的dyrk1a基因座)、或肌动蛋白测试f和g2r(用于修饰的β-肌动蛋白基因座)使pcr产物经受第二轮pcr扩增(表3)。对于各第二pcr反应,使0.1-1μl的第一pcr反应的pcr产物、0.5μl的正向引物(10μmol/μl)、0.5μl反向引物(10μmol/μl)、以及25μl的2xtaqpcrstarmix(genstar)和21-21.9μlh2o混合。pcr程序如下:96℃下3min;30个循环,94℃下30秒,57℃下30秒,并且72℃下20秒;72℃下5分钟。pcr产物用pcr纯化试剂盒纯化。然后将pcr产物克隆到t载体中并测序。
测序分析确认在ngago-gdna切割之后,供体egfpdna正确的插入了dyrk1a基因座中。seqidno:156是具有修饰的dyrk1a基因座的克隆的示例性序列。该克隆的测序色谱图的部分示于图21b-21d,包括靠近dy2引物结合区的上游序列(图21b)、dyrk1a与egfp序列起始部之间的5′接头(图21c),以及围绕g2r引物结合区域的下游序列(图21d)。在dyrk1a-egfp处,由于ngago-gdna的切割和核苷酸移除,接头是突变的g10序列(图21c,顶部)。另外,共转染后一个月的细胞荧光显微镜检查(图21e)示出表达egfp的细胞菌落。phago-gdna、miago-gdna和maago-gdna也有效地将供体egfpdna敲入dyrk1a基因座中,如由细胞中egfp的表达所指示(图23a、23c和23d)。
此外,显微镜法(图22a-22b)和测序分析(图22c)表明,ngago-g21gdna有效地将egfp供体dna敲入β-肌动蛋白基因座中。phago-gdna也有效地将供体egfpdna敲入β-肌动蛋白基因座中,如由细胞中egfp和f-肌动蛋白的共表达所指示(图23b)。
实施例7:实验方法和核酸序列
实施例1-6中的实验结果部分报道于gaof.等人“dna-guidedgenomeeditingusingthenatronobacteriumgregoryiargonaute,”nat.biotechnol.34:768-773(2016)中,其内容全文以引用方式并入本文。以下示例性方案涉及293t细胞中的ngago-gdna。可修改方案以在感兴趣的任何细胞系中评估其它ago-gdna系统。
ago-gdna介导的靶修饰
细胞培养物:使293t(atcccrl-3216)、hela(atccccl-2)、mcf7(atcchtb-22)细胞维持于高葡萄糖dulbecco改良的eagle培养基(dmem,hyclone)中,并使k562细胞(atccccl-243)维持于prmi-1640培养基中。在37℃和5%co2下,使培养基补充10%fbs(hyclone)、100u/ml青霉素和100μg/ml链霉素。在转染前8小时,将细胞接种至24孔板(costar),汇合度为60%。在转染前三十分钟,将细胞用磷酸盐缓冲盐水(pbs)洗涤两次,并将培养基更换成含有2%fbs的高葡萄糖dmem培养基。对培养基进行加热灭活,以除去微生物如支原体、衣原体、古生菌、原生动物和真菌的污染。
gdna的制备:单链dna寡核苷酸可商购获得。为制备5′磷酸化引导dna,如下使用t4pnk(biolab)使ssdna寡核苷酸5′磷酸化。通过使2μlt4pnk、12μlt4连接酶缓冲液(包含atp)、100μlssdna(约33μg)的水溶液与6μl水混合来制备120μl反应混合物。如果使用不含atp的t4连接酶缓冲液,则将2μl25mmatp添加到反应混合物中,并改为添加4μl水至120μl的最终体积。将反应混合物在37℃下温育过夜。在5′磷酸化之后,可直接使用所得的gdna而无需纯化。将gdna用水(ph8)稀释至300μl,以实现10nm或100ng/μl的最终浓度。另选地,5′磷酸化单链寡核苷酸引导dna可商业购买。
转染:使用
为了整合供体dna,进行两次转染。在第一转染中,在24孔板的各个孔中,将300ngnls-ngago质粒和300nggdna用50μlopti-mem(gibco)进行稀释,并将1.25μl脂质转染胺2000用50μlopti-mem进行稀释。使dna混合物和脂质转染胺混合物保持5min。然后通过轻轻抽吸混合dna混合物和脂质转染胺混合物,并温育20分钟。然后将dna/脂质转染胺混合物添加至各个孔的细胞。在第一转染后20小时,进行第二转染。在第二转染前三十分钟,将细胞用pbs洗涤两次,并将培养基更换成含有2%fbs的高葡萄糖dmem培养基。对于24孔板的各个孔,将1μg供体dna用50μlopti-mem(gibco)进行稀释,并将1.25μl
因为ngago遵循“一种-引导物-忠实性”规则(即引导物仅可在ngago蛋白处于表达过程时被负载)以提高gdna负载于ngago的效率,可对gdna进行多次转染(例如,在初次转染后8、12或24小时)。在收获之前,将细胞温育48-60小时。在收获时90%的细胞汇合度是理想的。铺板中细胞的显著减弱了基因编辑的功效。例如,发现hdr介导的编辑仅在s和g2期发生。此处不应使用脂质转染胺3000,原因是p3000干扰ssdna转染。
修饰基因座的基因组dna提取和pcr扩增
基因组dna提取:在转染后48-60小时(细胞的理想汇合度为90%),通过胰蛋白酶消化收获细胞。将四个孔中的细胞合并到1.5mleppendorf管中。对于基因组dna提取,将500μl细胞裂解缓冲液(50mmtris,100mmedta,0.5%sds,ph8)和10μl蛋白酶k(10mg/ml)加入到各个管中并轻轻混合。将管在55℃水浴中温育2小时。对于酚-氯仿提取,将200μltris-酚和200μl三氯甲烷添加到各个管中,并且轻轻而充分地混合。在温育5小时后,将样品以12,000rpm旋转15分钟,以使水相与酚相分离。将水相小心地收集到干净的eppendorf管中。将酚-氯仿提取重复一次并合并水相。将500μl三氯甲烷添加到收集的水相中,轻轻而充分地混合。在温育5分钟后,将样品以12,000rpm旋转15分钟,以使水相与酚相分离。将水相小心地移取到干净的eppendorf管中。将整个酚-氯仿萃取重复一次。为了沉淀dna,将900μl乙醇添加到收集的水相中并在-20℃下温育30分钟。将样品以12,000rpm离心10分钟,并且将dna沉淀用500μl75%乙醇洗涤三次。使dna沉淀空气干燥,并加入50μltris-edta缓冲液。将基因组dna浓度调节至100ng/μl。
pcr扩增:对于每20μl的pcr反应,将1μl的基因组dna、0.5μl的正向引物(10μmol/μl)、0.5μl反向引物(10μmol/μl)、以及10μl的2xtaqpcrstarmix(genstar)和8μlh2o混合。pcr程序如下:96℃下3min;30个循环,94℃下30秒,57℃下30秒,并且72℃下20秒;72℃下5分钟。pcr产物用pcr纯化试剂盒纯化。
t7ei测定
哺乳动物细胞基因组中的双链断裂主要通过内源性非同源末端连接(nhej)途径进行修复,导致插入缺失。t7内切核酸酶i(t7ei)酶识别并切割未完全匹配的dna,诸如插入缺失。因此,下文所述的t7ei测定可用于确定nejh引入的插入缺失率,这与ago-gdna系统或cas9-sgrna系统的双链断裂率相关。
t7ei反应:使纯化的pcr产物变性,再退火,并用t7内切核酸酶i消化。使150ng的pcr产物与1μl10xnebuffer2(newenglandbiolabs)、以及ddh2o混合至9.6μl的最终体积。然后将混合物在95℃下温育3min,继之以95~85℃-2℃/秒,然后是85~25℃-0.1℃/秒的温度梯度。接着将样品在4℃下温育1小时。将9.6μl退火产物与0.4μlt7ei(newenglandbiolabs)混合,并在37℃下温育30分钟。将10μl的t7ei反应产物与2μl6x上样缓冲液(biolab)混合,使其在140v下经受变性page电泳30分钟,并利用银染可视化。
使用imagelab(bio-rad)分析page凝胶上的条带强度。切割效率(即插入缺失百分比)可由化学式(1-(1-(b+c/a+b+c))1/2)×100确定,其中a为dna底物的条带强度,并且b和c为切割产物的条带强度。
变性的page和核酸的银染
变性的聚丙烯酰胺凝胶电泳(page)凝胶(1mm梳板)采用含有4m尿素、1.5ml的5xtris-硼酸edta缓冲液(tbe)、85μl的10%过硫酸铵(aps)、3.8μl的四甲基乙二胺(temed)、以及4ml的ddh20的1ml30%(w/v)双-丙烯酰胺(19:1)溶液进行制备。使用
在聚丙烯酰胺凝胶电泳之后,使用银染来可视化核酸。在电泳之后,将page凝胶在轻柔振荡下直接置入fix溶液(10%乙酸于milliq水中)中30min。然后将凝胶用超纯milliq水洗涤三次,每次4min,接着在振荡下于冰冷却的染色溶液(0.1g硝酸银和150μl甲醛溶于100mlmilliq水)中染色25min。在染色之后,将凝胶用milliq水洗涤三次(每次10秒)并在轻柔振荡下置入冰冷却的显影液(6g碳酸钠,300μl甲醛和13μl30%硫代硫酸钠溶于200mlmilliq水)中直至明显出现条带。然后倾析出显影液并加入固定溶液以终止反应。
核酸序列
用于实施例1-6中的引导dna的序列在表2中列出。pcr引物,包括t7ei测定所用的那些列于表3中。其它寡核苷酸、质粒和构建体示于表4中。
表2:引导dna:
表3:pcr引物
表4:其它寡核苷酸、质粒和构建体:
实施例8:argonaute间的序列同源性
通过使用嗜热栖热菌argonaute(“ttago”)和强烈火球菌(pyrococcusfuriosus)argonaute(“pfago”)的氨基酸序列,针对美国国家生物技术信息中心(ncbi)非冗余蛋白序列数据库执行位置特异性迭代基本局部比对搜索工具(psi-blast),鉴别得自格氏嗜盐碱杆菌sp2的argonaute(ngago,蛋白质标识符afz73749.1)、phago和miago。通过使用ngago、phago和miago的氨基酸序列执行psi-blast类似地鉴别本文所述的其它argonaute。
表1中的argonaute都能够在10-60℃(尤其是37℃)下修饰dna。这些argonaute能够使用5′磷酸化单链引导dna来切割靶dna的两条链,从而诱导双链断裂。相比之下,来自大部分其它菌种的argonaute利用rna引导物来切割靶rna。
图24a和24b示出了围绕5′磷酸盐结合位点和核酸酶活性位点的ago蛋白的序列比对。本文所述的大部分ago蛋白在mid结构域中5′-磷酸盐结合位点的kqk基序中包含3个保守氨基酸中的至少2个(图24a中突出显示);并在piwi结构域中核酸酶活性位点的dde基序中包含3个保守氨基酸中的至少2个(图24b中突出显示)。图25示出ago蛋白基于其序列同源性的系统发育树。
实施例9:细菌中表达的ngago-gdna对靶dna的降解
据报道,细菌和古生菌中存在的5′端磷酸化短dna,尽管5′端磷酸化机理尚不清楚。该实施例展示ngago在细菌细胞中结合侵染性dna并降解靶dna基因组的能力。
ngago-gdna的体内基因组降解
通过用包含ngago表达盒的线性化pgex-6p-1质粒(ngago-6p-1,图5)转化大肠杆菌mg1655,制备稳定转基因ngago-表达大肠杆菌菌株(大肠杆菌mg1655-ngago)。ngago盒受lac操纵子的控制,其可通过在培养物中添加iptg来诱导。
在图26a的组1中,将大肠杆菌mg1655-ngago细菌铺板到lb平板上,其在平板右手侧补充有0.1mmiptg。转基因细菌在平板左侧和右侧均存活。在图26a的组2中,将大肠杆菌mg1655-ngago细菌用不相关gfp-n1质粒(与ngago-6p-1质粒无序列同源性)转化,并铺板到在右手侧补充有0.1mmiptg的lb平板上。同样,转基因细菌在平板左侧和右侧均存活。在图26a的组3中,将大肠杆菌mg1655-ngago细菌用pgex-6p-1质粒转化,并铺板到lb平板上。因为pgex-6p-1序列处于大肠杆菌mg1655-ngago细菌的基因组中,在平板右侧添加0.1mmiptg后,细菌死亡。不受任何理论或假设的约束,细菌细胞表达的ngago结合了源于降解的pgex-6p-1质粒的gdna,并且ngago-gdna复合物靶向细菌细胞的基因组并使之断裂。
ago-gdna的体外靶dna降解
用包含融合有gst标签的ngago、miago或maago表达盒的pgex-6p-1质粒转化大肠杆菌菌株bl21de3。使用gst纯化所表达的ago蛋白与其结合的引导dna,并与线性化空载体pgex-6p-1一起温育,随后通过琼脂糖凝胶电泳进行分析。
简而言之,使用ngago作为实施例,使ngago-6p-1质粒转化到大肠杆菌菌株bl21de3中,以提供ngago-6p-1bl21细菌菌株。将5mllb培养基(补充有1∶1000氨苄青霉素)以1∶1000比率接种有ngago-6p-1bl21,并温育过夜。将过夜的细菌培养物以1∶100比率接种到50ml培养基中,并在200rpm和37℃下温育3小时,直至od600达到0.8-1.0。将细菌培养物和摇动器冷却至21℃,并将0.1mmiptg和20mmmgcl2添加到培养物中,以在振荡和100rpm下诱导细菌。在诱导8小时后收获细菌,并在-20℃储存细菌沉淀。
为了从细菌沉淀纯化ngago与其所缔合的gdna,将细菌沉淀从-20℃冷冻机中移除,在冰上解冻,并重悬于20ml预冷却的裂解缓冲液中(20mmtris-hcl,ph7.0,20mmmgcl2,100mmnacl)。然后将混合物在250w下超声处理,3秒启动,8秒断开,共10分钟。随后将超声处理的混合物在4℃,12000rpm下超声处理45分钟。将上清液与20μlgst琼脂糖珠一起温育3小时,并允许上清液流动通过。将gst珠用50ml的洗涤缓冲液(20mmtris-hcl,ph7.0,20mmmgcl2,100mmnacl)洗涤3次。然后用洗脱缓冲液(20mmtris-hcl,ph7.0,20mmmgcl2,100mmnacl,20mm还原谷胱甘肽)洗脱ngago。因为原核生物具有内源性ssdna产生机制,纯化的ngago预负载有ssdna。
体外酶切割反应如下设定:使200ng纯化的ngago与100ng线性化pgex-6p-1、1μl10x缓冲液(10mmtris-hcl,ph7.0,150mmnacl,1mmmgcl2,0.4%甘油和20μg/mlbsa)、以及水混合至10μl的总体积。将酶切割反应混合物在37℃下温育4小时或8小时,然后经受1%琼脂糖凝胶电泳和银染。
图26b示出体外酶反应混合物的电泳结果。因为ago与来源于大肠杆菌中pgex-6p-1质粒的gdna结合,ago-gdna复合物可在体外完全降解线性化pgex-6p-1空载体。
- 还没有人留言评论。精彩留言会获得点赞!