一种鉴定甜樱桃啤酒花矮化类病抗性的方法与流程
本发明属于生物技术领域,具体涉及一种鉴定甜樱桃啤酒花矮化类病抗性的方法。
背景技术:
啤酒花矮化类病毒 (Hop stunt viroid,HSVd)属于马铃薯纺锤块茎类病毒科(Pospiviroidae),是啤酒花矮化类病毒属 (Hostnviroid)的唯一成员。其寄主范围非常广泛,包括啤酒花、黄瓜、葡萄、柑橘、梨、桃、李、杏和扁桃等。其中葡萄和杏在感染HSVd后不表现症状,而啤酒花、桃、李和柑橘被感染后则会引发严重的病害,如啤酒花矮化、李和桃斑果病、柑橘矮化和裂皮病等。
类病毒内及类病毒间的重组现已被认为对类病毒的起源进化具有重要意义且与类病毒致病性密切有关,而类病毒侵染性克隆技术是研究类病毒与寄主互作分子机制的有效手段。构建类病毒侵染性克隆已成为国际上开展类病毒生物学活性、序列变异、致病机理等相关研究的重要实验手段,因此构建啤酒花矮化类病毒侵染性克隆,将对深入研究该类病毒序列与功能之间的关系具有重要意义。现有技术中,针对甜樱桃啤酒花矮化类病毒克隆的构建等方面还未曾有报道。
技术实现要素:
针对现有技术中存在的问题,本发明提供了一种鉴定甜樱桃啤酒花矮化类病抗性的方法。
本发明所采用的具体的技术方案为:
本发明提供了一种鉴定甜樱桃啤酒花矮化类病抗性的方法,包括以下步骤:
(1)植物总RNA的提取及引物设计:提取感病甜樱桃叶片总RNA, -80℃保存备用;
(2)HSVd全长cDNA克隆及测序:以提取的总RNA为模板,反转录试剂盒中的随机引物进行RT-PCR反转录,反应体系为20μL;以反转录得到的cDNA为模板进行PCR扩增反应,R1和F1为引物;
PCR产物经1.5%琼脂糖凝胶电泳,用快速琼脂糖凝胶DNA回收试剂盒(康为) 回收目的片段,将回收的片段与pGEM-T(Promega)载体连接,连接产物转化大肠杆菌DH5α感受态细胞,菌液PCR筛选阳性质粒;
(3)HSVd侵染性克隆的构建:将所得分离物序列与HSVd序列进行比较,对测序正确的菌株摇菌提取质粒;以提取的质粒为模板,以4个HSVd全长片段的HSVd-F1/HSVd-R1和HSVd-F2/HSVd-R2为引物,引物对HSVd-F1/HSVd-R1用于扩增第一个片段,引物对HSVd-F2/HSVd-R2用于扩增第二个片段,利用高保真酶分别进行PCR扩增获得HSVd全长片段,电泳后切胶回收;将两个片段与载体进行重组反应,反应结束后瞬时离心,将离心管置于冰上冷却5min;重组产物转化大肠杆菌DH5α感受态细胞,菌液PCR筛选阳性质粒;
(4)质粒接种及抗病性鉴定:将获得含有HSVd cDNA加倍串联序列的克隆质粒pGEM-HSVd-HSVd,提取质粒pGEM-HSVd-HSVd,通过茎杆注射方法进行接种甜樱桃砧木G6,同时以接种空白质粒pGEM的植株为阴性对照。
进一步的,步骤(2)中,所述引物对序列R1和F1如SEQ ID NO:1和SEQ ID NO:2所示。
R1(5′-TTGTTCTATCTGCGCCTCTGCC-3′)和F1(5′-AAGCAGGTTGGAGCGAACCGA-3′)
进一步的,步骤(2)中,所述反转录的反应体系中含有总RNA 2μg,2×TS Reation Mix 10 μL,随机引物2 pmol,TransScriptRT/RI Enzyme Mix 1μL,加ddH2O至20 μL,混匀;42℃孵育30 min,冰上冷却2 min,-20℃保存备用。
进一步的,步骤(2)中,所述PCR扩增反应的体系为50μL,含有5×TransStart FastPfu Buffer 10μL,2.5 mM dNTPs 5μL,TransStart FastPfu DNA聚合酶1μL,引物对R1和F1各1μL (10μM),模板2μL,加ddH2O至50μL, 混匀;PCR扩增反应程序为94℃预变性5 min;94℃变性30 s,52℃退火30 s,72℃延伸30 s,35个循环;最后72℃延伸10 min。
进一步的,所述第一个片段的上游引物HSVd-F1如SEQ ID NO:3所示;(CGCGGGAATTCGATTGCAACTCTTCTCAGAATCCAG),所述下游引物HSVd-R1如SEQ ID NO:4所示(GAAGAGTTGCCCCGGGGCTCCTTTC);所述第二个片段的上游引物HSVd-F2如SEQ ID NO:5所示(CCCCGGGGCAACTCTTCTCAGAATC),所述下游引物HSVd-R2如SEQ ID NO:6所示(AATTCACTAGTGATTCCCGGGGCTCCTTTCTCAGG)。
进一步的,步骤(3)中,所述重组反应的体系为,pGEM-T线性化载体0.25 pmol,两个插入片段(总量)0.5 pmol,2×EasyGeno Assembly Mix 5μL,加ddH2O补足至10μL;将混合反应液置于50℃反应15 min。
本发明的有益效果为:本发明成功构建了甜樱桃HSVd的侵染性克隆,进行人工接种进行抗病性鉴定。本发明克服了田间多种病毒复合侵染症状不单一,类病毒毒源保存等技术难度,构建了高效引发甜樱桃类病毒病的人工接种鉴定方法。该方法操作简单、效率高、重复性和可控性好。能提供相对一致的接种压力,是一种简单有效的接种鉴定方法。生物学活性分析显示本方法所获得的HSVd串联重复cDNA克隆能成功地侵染甜樱桃砧木G6,表明该克隆具有侵染活性,可用于相关类病毒功能基因研究和植物抗病性分析。
附图说明
图1为实施例1中质粒接种后的甜樱桃砧木。
其中,a为健康植株、b为质粒接种后的发病植株。
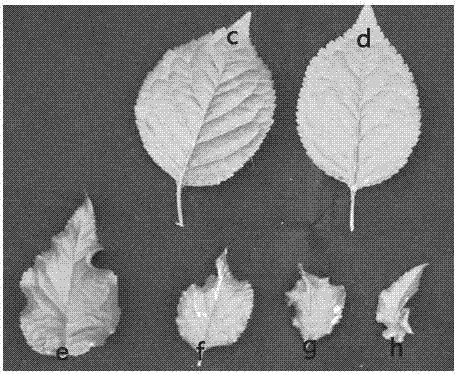
图2为质粒接种后甜樱桃叶片。
其中,c、d为健康叶片,e-h为发病叶片。
具体实施方式
下面通过实施例对本发明进行进一步的阐述,应该明白的是,下述说明仅是为了解释本发明,并不对其内容进行限定。
实施例1
1.1植物总RNA的提取及引物设计
用EASYspin Plus植物RNA提取试剂盒提取感病甜樱桃叶片总RNA,具体方法参照试剂盒说明书(AidLab, China),用NanoDrop2000型紫外分光光度计检测RNA纯度,并用1.5%琼脂糖凝胶电泳检测RNA的完整性,-80℃保存备用。
HSVd扩增引物根据Genbank中登录的HSVd的RNA序列(Acc.NO. KJ754184.1)设计了两条引物,引物R1(5′-TTGTTCTATCTGCGCCTCTGCC-3′)和F1(5′-AAGCAGGTTGGAGCGAACCGA-3′) 用于PCR扩增,引物由上海生工生物有限公司设计合成;
1.2、HSVd全长cDNA克隆及测序
以提取的总RNA为模板,反转录试剂盒(TransGen Biotech)中的通用引物进行RT-PCR反转录,体系为20 μL;反转录反应体系中含有总RNA 2μg,2×TS Reation Mix 10 μL,随机引物2 pmol,TransScriptRT/RI Enzyme Mix 1μL,加ddH2O至20 μL,混匀;42℃孵育30 min,冰上冷却2 min,-20℃保存备用;
以反转录得到的cDNA为模板进行PCR反应,R1和F1为引物。PCR扩增体系为50μL,含有5×TransStart FastPfu Buffer 10μL,2.5 mM dNTPs 5μL,TransStart FastPfu DNA聚合酶1μL,引物对R1和F1各1μL (10μM),模板2μL,加ddH2O至50μL, 混匀;PCR反应程序为94℃预变性5 min;94℃变性30 s,52℃退火30 s,72℃延伸30 s,35个循环;最后72℃延伸10 min;
PCR产物经1.5%琼脂糖凝胶电泳,用快速琼脂糖凝胶DNA回收试剂盒(康为) 回收目的片段,将回收的片段与pGEM-T(Promega)载体连接,连接产物转化大肠杆菌DH5α感受态细胞,菌液PCR筛选阳性质粒,送交上海生工生物有限公司测序;
测序结果已在GenBank中注册,注册号为 KX355200;序列如下:
GCAACTCTTCTCAGAATCCAGCGAGAGGCGTGGAGAGAGGGCCGCGGTGCTCTGGAGTAGAGGCTCTGCCTTCGAAACACCATCGATCGTCCCTTCTTCTTTACCTTCTTCTGGCTCTTCTTGGAGACGCGACCGGTGGCACCCCTGCTCGGTTCGCTCCAACCTGCTTTTGTTCTATCTGCGCCTCTGCCGCGGATCCTCTCTTGAGCCCCTCTGGGGAATTCTCGAGTTGCCGCAAAAGGCATGCAAAGAAAAAAACTAGGCAGGGAGGCGCTTACCTGAGAAAGGAGCCCCGGG
1.3、HSVd侵染性克隆的构建
采用DNAMAN V.6.0软件将所得分离物序列与GenBank中已报道HSVd序列进行比较,对测序正确的菌株摇菌提取质粒;为将HSVd 全长片段以串联重复形式插入到载体pGEM-T(Promega)中,根据EasyGeno重组克隆试剂盒说明书设计4个HSVd全长片段的PCR引物,第一个片段的上游引物HSVd-F1(CGCGGGAATTCGATTGCAACTCTTCTCAGAATCCAG),下游引物HSVd-R1(GAAGAGTTGCCCCGGGGCTCCTTTC);第二个片段的上游引物HSVd-F2(CCCCGGGGCAACTCTTCTCAGAATC),下游引物HSVd-R2(AATTCACTAGTGATTCCCGGGGCTCCTTTCTCAGG)。以提取的质粒为模板,HSVd-F1/HSVd-R1和HSVd-F2/HSVd-R2为引物,利用高保真酶分别进行PCR扩增获得HSVd全长片段,电泳后切胶回收;将两个片段与载体进行重组反应,反应体系为,pGEM-T线性化载体0.25pmol,两个插入片段(总量)0.5pmol,2×EasyGeno Assembly Mix 5μL,加ddH2O补足至10μL。将混合反应液置于50℃反应15min,反应结束后瞬时离心,将离心管置于冰上冷却5min;重组产物转化大肠杆菌DH5α感受态细胞,菌液PCR筛选阳性质粒,送交生上海生工生物有限公司测序;
1.4、质粒接种及抗病性鉴定
经过对重组质粒的筛选鉴定及测序分析,获得含有HSVd cDNA加倍串联序列的克隆质粒pGEM-HSVd-HSVd;提取质粒pGEM-HSVd-HSVd,通过茎杆注射方法进行接种,同时以接种空白质粒pGEM的植株为阴性对照;接种30 d后发现接种pGEM-HSVd-HSVd质粒的甜樱桃砧木出现叶片翻卷、不平整等症状,而阴性对照的砧木上则无任何明显症状表现。
对比例1
1.1植物总RNA的提取及引物设计
用EASYspin Plus植物RNA提取试剂盒提取感病甜樱桃叶片总RNA,具体方法参照试剂盒说明书(AidLab, China),用NanoDrop2000型紫外分光光度计检测RNA纯度,并用1.5%琼脂糖凝胶电泳检测RNA的完整性,-80℃保存备用。
HSVd扩增引物根据Genbank中登录的HSVd的RNA序列(Acc.NO. KJ754184.1)设计了两条引物,引物R2(5′-AACCTGCTTTTGTTCTATCTG -3′)和F2(5′-AACCTGCTTTTGTTCTATCTG -3′) 用于PCR扩增,引物由上海生工生物有限公司设计合成;
1.2、HSVd全长cDNA克隆及测序
以提取的总RNA为模板,反转录试剂盒中的通用引物进行RT-PCR反转录,体系为20 μL;反转录反应体系中含有总RNA 2μg,2×TS Reation Mix 10 μL,随机引物2 pmol,TransScriptRT/RI Enzyme Mix 1μL,加ddH2O至20 μL,混匀;42℃孵育30 min,冰上冷却2 min,-20℃保存备用;
以反转录得到的cDNA为模板进行PCR反应,R2和F2为引物。PCR扩增体系为50μL,含有5×TransStart FastPfu Buffer 10μL,2.5 mM dNTPs 5μL,TransStart FastPfu DNA聚合酶1μL,引物对R2和F2各1μL (10μM),模板2μL,加ddH2O至50μL, 混匀;PCR反应程序为94℃预变性5 min;94℃变性30 s,52℃退火30 s,72℃延伸30 s,35个循环;最后72℃延伸10 min;
PCR产物经1.5%琼脂糖凝胶电泳,检测不到目的条带。
对比例2
2.1植物总RNA的提取及引物设计
用EASYspin Plus植物RNA提取试剂盒提取感病甜樱桃叶片总RNA,具体方法参照试剂盒说明书(AidLab, China),用NanoDrop2000型紫外分光光度计检测RNA纯度,并用1.5%琼脂糖凝胶电泳检测RNA的完整性,-80℃保存备用。
HSVd扩增引物根据Genbank中登录的HSVd的RNA序列(Acc.NO. KJ754184.1)设计了两条条引物,引物R3(5′-CTCAGAATCCAGCGAGAG -3′)和F3(5′-AGGCGTGGAGAGAGGGC -3′) 用于PCR扩增,引物由上海生工生物有限公司设计合成;
2.2、HSVd全长cDNA克隆及测序
以提取的总RNA为模板,反转录试剂盒中的通用引物进行RT-PCR反转录,体系为20 μL;反转录反应体系中含有总RNA 2μg,2×TS Reation Mix 10 μL,随机引物2 pmol,TransScriptRT/RI Enzyme Mix 1μL,加ddH2O至20 μL,混匀;42℃孵育30 min,冰上冷却2 min,-20℃保存备用;
以反转录得到的cDNA为模板进行PCR反应,R3和F3为引物。PCR扩增体系为50μL,含有5×TransStart FastPfu Buffer 10μL,2.5 mM dNTPs 5μL,TransStart FastPfu DNA聚合酶1μL,引物对R3和F3各1μL (10μM),模板2μL,加ddH2O至50μL, 混匀;PCR反应程序为94℃预变性5 min;94℃变性30 s,52℃退火30 s,72℃延伸30 s,35个循环;最后72℃延伸10 min;
PCR产物经1.5%琼脂糖凝胶电泳,检测不到目的条带。
<110>山东省果树研究所
<120>一种鉴定甜樱桃啤酒花矮化类病抗性的方法
<160>6
<210>1
<211>22
<212>DNA
<213>人工合成
<400>1
TTGTTCTATC TGCGCCTCTG CC 22
<210>2
<211>21
<212>DNA
<213>人工合成
<400>2
AAGCAGGTTG GAGCGAACCG A 21
<210>3
<211>36
<212>DNA
<213>人工合成
<400>3
CGCGGGAATT CGATTGCAAC TCTTCTCAGA ATCCAG 36
<210>4
<211>25
<212>DNA
<213>人工合成
<400>4
GAAGAGTTGC CCCGGGGCTC CTTTC 25
<210>5
<211>25
<212>DNA
<213>人工合成
<400>5
CCCCGGGGCA ACTCTTCTCA GAATC 25
<210>6
<211>35
<212>DNA
<213>人工合成
<400>6
AATTCACTAG TGATTCCCGG GGCTCCTTTC TCAGG 35
- 还没有人留言评论。精彩留言会获得点赞!