茶氨酰氨基酸苄酯修饰的姜黄素,其合成,活性和应用的制作方法
本发明涉及1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮,涉及它们的制备方法,涉及它们的抗肿瘤生长活性,涉及它们的逆转肿瘤细胞耐药活性,以及涉及它们的抗炎活性,因而本发明涉及它们在制备抗肿瘤药物,逆转肿瘤细胞耐药药物和抗炎药物中的应用。本发明属于生物医药领域。
背景技术
恶性肿瘤是严重危害人类健康的全球性问题。肿瘤患者发现病患时大多已是临床中晚期,治疗方法以放化疗为主,化疗是恶性肿瘤处于中晚期时的主要治疗手段。然而,化疗过程中不仅肿瘤细胞对抗肿瘤药物产生耐药性严重影响化疗效果,而且炎症也会进一步恶化肿瘤患者的预后。由于现有抗肿瘤药物既没有逆转肿瘤细胞耐药作用也没有抗炎作用,所以肿瘤化疗的临床疗效不理想。发明具有抗炎活性并可逆转肿瘤细胞耐药的抗肿瘤药物是临床的迫切需求。此前,发明人曾公开1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰氨基酸苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮具有明显的抑制肿瘤细胞增殖的活性。后来发明人又公开抗黏附肽修饰的姜黄素在1μmol/kg剂量下可抑制s180小鼠肿瘤生长和抑制icr小鼠的炎症。可是最低有效剂量为1μmol/kg。为了降低最低有效剂量,发明人对姜黄素的酚羟基展开了各种修饰。经过3年探索,发现用茶氨酰-aa-obzl(aa选自l-asp残基和l-glu残基)修饰的姜黄素不仅可使抗肿瘤生长的最低有效剂量降至0.1μmol/kg,而且抗炎的最低有效剂量也低至0.1μmol/kg。此外,化合物同时具有逆转耐药的活性。抗肿瘤生长的有效剂量降低10倍表明,加上逆转耐药和抗炎作用说明这种结构修饰有突出的技术效果。根据这些发现,发明人提出了本发明。
技术实现要素:
本发明的第一个内容是提供下式所示的1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(式中aa选自l-asp残基和l-glu残基)。
本发明的第二个内容是提供1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(式中aa选自l-asp残基、l-glu残基和l-phe残基)的合成方法,该方法包括:
(1)制备6-(4-羟基-3-甲氧苯基)-5,6-己烯-2,4-二酮(1);
(2)制备2-(4-甲酰基-2-甲氧基苯氧基)-乙酸苄酯(2);
(3)以步骤(1)和步骤(2)的产物为反应原料制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(3);
(4)将1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮皂化,得到1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰乙酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(4);
(5)化合物4和l-茶氨酸苄酯偶联得到1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(5);
(6)将化合物5皂化得到1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(6);
(7)化合物6和l-氨基酸苄酯偶联得到1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮。
本发明的第三个内容是评价1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮对s180小鼠肿瘤生长的抑制应用。
本发明的第四个内容是评价1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮对耐药的肿瘤细胞逆转耐药的应用。
本发明的第五个内容是评价1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮对icr小鼠炎症的抑制作用。
附图说明
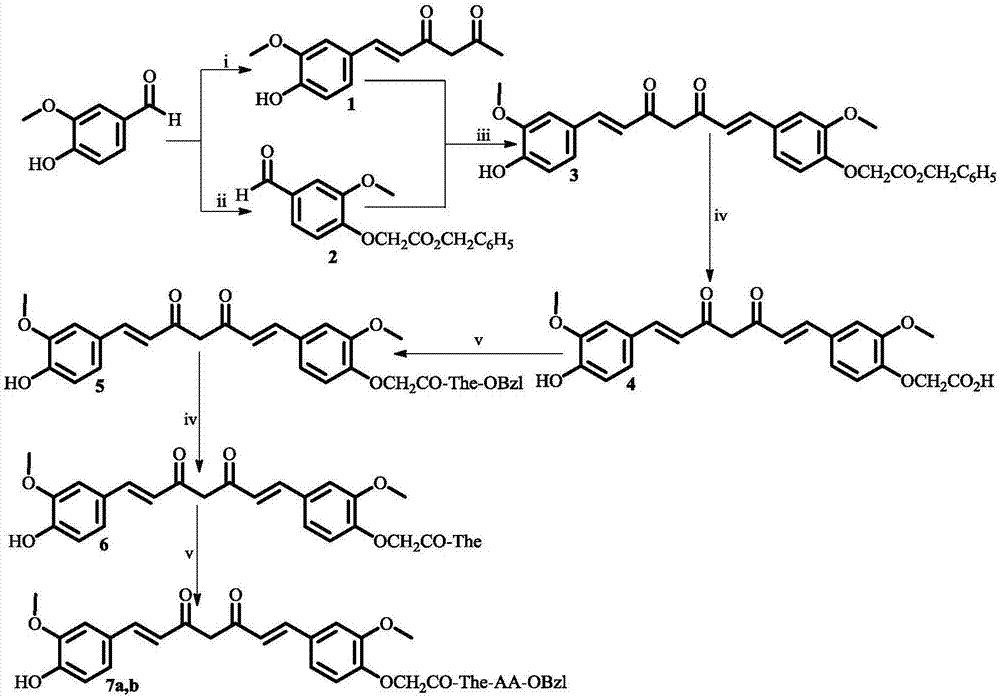
图1为1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-aa-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮.7a中aa为l-asp残基;7b中aa为l-glu残基;i)三氧化二硼(b2o3),乙酰丙酮,硼酸三正丁酯,正丁胺,氯化氢水溶液(1m);ii)碳酸钾,溴乙酸苄酯;iii)三氧化二硼(b2o3),硼酸三正丁酯,正丁胺,10%乙酸水溶液;iv)氢氧化钠水溶液(2m),丙酮;v)二环己基碳二亚胺(dcc),1-羟基苯并三唑(hobt),n-甲基吗啉(nmm),四氢呋喃(thf)。
具体实施方式
为了进一步阐述本发明,下面给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1制备6-(4-羟基-3-甲氧苯基)-5,6-己烯-2,4-二酮(1)
将45.0ml(437.7mmol)乙酰丙酮,21.0g(301.6mmol)氧化硼和150.0ml无水乙酸乙酯于60℃回流1h。然后,往里加22.5g(148.0mmol)香草醛和41ml(293.0mmol)硼酸三丁酯。反应混合物70℃搅拌30min。于30min内继续往里加15ml(205.1mmol)正丁胺与135ml乙酸乙酯中的溶液。混合物于100℃搅拌3h后冷却至室温,并往里滴加150ml盐酸(1m)。混合物于50℃搅拌30min,静置,充分分层,水层用乙酸乙酯萃取3次。合并的乙酸乙酯层用饱和nacl溶液洗至中性,无水硫酸钠干燥,过滤,滤液减压浓缩至干,残留物用硅胶柱层析纯化(石油醚/乙酸乙酯=4/1)得到10.05g(29%)目标化合物,为黄色固体。esi-ms(m/e):235[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=15.74(s,1h),9.64(s,1h),7.50(d,j=15.9hz,1h),7.30(s,1h),7.12(d,j=8.1hz,1h),6.82(d,j=8.1hz,1h),6.64(d,j=15.9hz,1h),5.84(s,1h),5.14(s,2h),3.83(s,3h),2.12(s,3h)。
实施例2制备3-甲氧基-4-(氧基-2-乙酰苄酯基)苯甲醛(2)
将10g(65.8mmol)香草醛溶于100ml无水四氢呋喃。向该溶液分批加入10.9g(79.0mmol)碳酸钾并搅拌3h。然后向溶液中滴加9.3ml溴乙酸苄酯,室温搅拌48h,tlc监(石油醚/乙酸乙酯=3/1)显示反应结束。反应混合物过滤,滤液减压浓缩,残余物用100ml乙醚磨洗静置12h后倒掉乙醚,10ml乙醚磨洗3次,去乙醚,得到15.4g(78%)标题化合物,为无色固体。esi-ms(m/e):301[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=9.86(s,1h),7.50(dd,j1=8.4hz,j2=1.8hz,1h),7.44(d,j=1.8hz,1h),7.39(s,5h),7.11(d,j=8.4hz,1h),5.21(s,2h),5.03(s,2h),3.84(s,3h)。
实施例3制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(3)
将5.55g(23.7mmol)6-(4-羟基-3-甲氧苯基)-5,6-己烯-2,4-二酮(1),0.83g(11.9mmol)氧化硼和100ml乙酸乙酯的悬浮液于70℃回流1h。之后,减压浓缩。残留物用100ml无水dmf溶解。往得到的溶液中加10.67g(35.6mmol)3-甲氧基-4-(氧基-2-乙酰苄酯基)苯甲醛(2)和11.15ml(41.0mmol)硼酸三丁酯。得到的溶液于80℃搅拌30min。之后,往里分4次滴加0.98ml(6.4mmol)正丁胺,用时1h,得到的溶液于80℃继续搅拌3h。之后,往里加200ml预热至60℃的10%乙酸水溶液。得到的溶液于80℃继续搅拌1h。反应混合物冷却至室温,过滤,滤饼用柱层析纯化(石油醚/乙酸乙酯=3/1),得到6.63g(53%)标题化合物,为黄色固体。esi-ms(m/e):517[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=9.69(s,1h),8.51(d,j=7.5hz,1h),7.80(t,j=5.7hz,1h),7.59(d,j=3.0hz,1h),7.54(d,j=3.0hz,1h),7.37(m,7h),7.16(m,1h),6.89(m,4h),6.09(s,1h),5.14(s,2h),4.62(s,2h),4.37(m,1h),3.87(s,3h),3.84(s,3h),3.04(qd,j1=7.2hz,j2=5.7hz,2h),2.15(m,2h),2.05(m,1h),1.90(m,1h),0.98(t,j=7.2hz,3h)。
实施例4制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(4)
将5g(9.7mmol)1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(3)用丙酮溶解。室温下往里加naoh水溶液(2m),调节反应液ph为13并搅拌6h。tlc(石油醚/乙酸乙酯=3/1)显示反应完成。反应混合物用饱和khso4水溶液将ph调到7,减压浓缩,残余物用饱和khso4水溶液调ph到2。之后,用乙酸乙酯萃取3次。合并乙酸乙酯层,用饱和nacl溶液洗至中性,用无水硫酸钠干燥。过滤,滤液减压浓缩,残留物用无水乙醚磨洗,得到2.64g(64%)标题化合物,为红色固体。esi-ms(m/e):425[m-h]-;1hnmr(300mhz,dmso-d6):δ/ppm=9.55(s,1h),7.57(m,2h),7.37(m,2h),7.20(m,2h),6.79(m,4h),6.06(s,1h),4.74(s,2h),3.85(s,6h)。
实施例5制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(5)
冰浴下将2g(4.7mmol)1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(4),1.69g(5.6mmol)盐酸茶氨酸苄酯和0.761g(5.64mmol)n-羟基苯并三唑(hobt)用50ml无水四氢呋喃溶解。向溶液中滴入由1.16g(5.63mmol)二环己基羰二亚胺(dcc)和10ml无水四氢呋喃的溶液。反应液用n-甲基吗啉调ph到8,室温搅拌12h。tlc(石油醚/乙酸乙酯=3/1)显示反应完成。反应液减压浓缩至干,残余物用150ml乙酸乙酯溶解。过滤、滤液依次用饱和碳酸氢钠水溶液洗3次,饱和氯化钠水溶液洗3次,饱和硫酸氢钾水溶液洗3次,饱和氯化钠水溶液洗3次,饱和碳酸氢钠水溶液洗3次及饱和氯化钠水溶液洗3次。合并的乙酸乙酯层用无水硫酸钠干燥,过滤,滤液减压浓缩。残留物用柱层析(二氯甲烷/甲醇=80/1)纯化,得到2.24g(71%)标题化合物,为黄色固体。esi-ms(m/e):673[m+h]+;1hnmr(300mhz,dmso-d6):δ/ppm=9.69(s,1h),8.51(d,j=7.5hz,1h),7.80(t,j=5.7hz,1h),7.59(d,j=3.0hz,1h),7.54(d,j=3.0hz,1h),7.37(m,7h),7.16(m,1h),6.89(m,4h),6.09(s,1h),5.14(s,2h),4.62(s,2h),4.37(m,1h),3.87(s,3h),3.84(s,3h),3.04(qd,j1=7.2hz,j2=5.7hz,2h),2.15(m,2h),2.05(m,1h),1.90(m,1h),0.98(t,j=7.2hz,3h)。
实施例6制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(6)
将2.24g(3.3mmol)1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸苄酯基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(5)用丙酮溶解,室温下向溶液中滴加naoh水溶液(2m)至反应液ph为13,搅拌6h,tlc(石油醚/乙酸乙酯=3/1)显示反应完成。用饱和khso4水溶液将反应液ph调到7,减压浓缩,残余物用饱和khso4水溶液调ph到2。之后,用乙酸乙酯萃取3次,合并的乙酸乙酯层用饱和nacl水溶液洗至中性,用无水硫酸钠干燥,过滤,滤液减压浓缩得到1.07g(55%)标题化合物,为红色糖浆。esi-ms(m/e):581[m-h]-;1hnmr(300mhz,dmso-d6):δ/ppm=9.42(s,1h),8.1(m,1h),7.77(m,1h),7.57(m,1h),7.37(m,1h),7.24(m,2h),7.10(m,2h),6.96(d,j=5.1hz,1h),6.79(m,3h),6.06(s,1h),4.57(s,2h),4.20(m,1h),3.82(s,6h),3.01(m,2h),2.09(m,3h),1.77(m,1h),0.95(t,j=4.5hz,3h)。
实施例7制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-asp(obzl)-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(7a)
冰浴下将2.32g(3.98mmol)1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(6),1.67g(4.78mmol)hcl·asp(obzl)-obzl和0.65g(4.78mmol)n-羟基苯并三唑(hobt)用20ml无水四氢呋喃溶解。之后,往里滴加0.98g(4.78mmol)二环己基羰二亚胺(dcc)和5ml无水四氢呋喃的溶液。反应液用n-甲基吗啉(nmm)调ph为8,室温搅拌12h,tlc(二氯甲烷/甲醇=50/1)显示反应结束。反应液减压浓缩至干,残余物用150ml二氯甲烷溶解,抽滤、滤液依次用饱和碳酸氢钠水溶液洗3次,饱和氯化钠水溶液洗3次,饱和硫酸氢钾水溶液洗3次,饱和氯化钠水溶液洗3次,饱和碳酸氢钠水溶液洗3次及饱和氯化钠水溶液洗3次。合并的二氯甲烷层用无水硫酸钠干燥,过滤,滤液减压浓缩至干。残留物用柱层析纯化(二氯甲烷/甲醇=50/1),得到1.98g(56.7%)标题化合物,为黄色固体。mp:136-138℃;
实施例8制备1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酰-glu-obzl-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(7b)
采用实施例7的方法,从2.5g(5.87mmol)1-(4-羟基-3-甲氧基苯基)-7-(4-氧乙酰茶氨酸基-3-甲氧基苯基)-1,6-庚二烯-3,5-二酮(6)和3.59g(8.80mmol)hcl·glu(obzl)-obzl得到880mg(17.0%)标题化合物,为黄色固体。mp:140-142℃;
实施例9测定化合物7a,b的抗肿瘤生长活性
测定前将阿霉素,化合物6和化合物7a,b用生理盐水溶解,用于s180小鼠给药。在无菌环境中取接种于雄性icr小鼠10天生长旺盛的s180腹水瘤液,用生理盐水稀释成(1:2)的液体充分混合,将肿瘤细胞悬液用新鲜配制的0.2%台盼蓝染色,混匀后按白细胞计数方法计数,染蓝色者为死细胞,不染色者为活细胞。按细胞浓度=4大方格内活细胞数/4×104×稀释倍数=细胞数/ml计算细胞密度,按细胞存活率=活细胞数/(活细胞数+死细胞数)×100%计算细胞存活率。将存活率大于90%的瘤液用匀浆法制成密度为2.0×107个/ml的细胞悬液。该细胞悬液接种于小鼠右腋皮下(0.2ml/只),制造s180荷瘤小鼠。接种24h后s180荷瘤小鼠每日腹腔注射阿霉素的生理盐水溶液(剂量为2μmol/kg/天g)或腹腔注射化合物6的生理盐水溶液(剂量为1μmol/kg/天)或腹腔注射化合物7a,b的生理盐水溶液(剂量为0.1μmol/kg/天),每组10只。每天给药一次,连续给药12天。最后一次给药的次日乙醚麻醉脱颈椎处死,然后用镊子固定小鼠右腋肿瘤生长部位,剪开皮肤钝性剥离肿瘤并称重。用瘤重(均值±sdg)表示疗效,数据用t检验和方差分析。结果见表1。在0.1μmol/kg剂量下化合物7a,b不仅有效地抑制肿瘤生长,而且活性与剂量比它们大10倍的化合物6没有显著性差异。这些数据表明,本发明有显著的技术效果。
表1化合物7a,b对s180小鼠肿瘤生长的影响
a)与生理盐水比p<0.01,与化合物6比p<0.05;b)生理盐水组相比p<0.01,与化合物6比p<0.05,与阿霉素比p>0.05;n=10.
实施例10测定化合物7a,b逆转肿瘤细胞耐药的活性
测定前将阿霉素(adr),紫杉醇(tax),维拉帕米和化合物7a,b用dmso溶解,培养液稀释到所需浓度(dmso含量小于0.5%)用于给药。采用mtt法测定。维拉帕米是公认的具有逆转肿瘤细胞耐药活性的工具药,这里用作阳性对照。
将脱药3天生长状态良好,处于对数生长期的紫杉醇耐药的贴壁细胞a549/tax以6×104个/ml的密度接种于96孔板,每孔100μl。在5%co2培养箱中37℃孵育6小时。化合物孔先加25μl浓度为10μm的化合物7a,b,然后按预设浓度加25μl紫杉醇溶液。空白对照孔先加50μl溶媒,然后按预设浓度加25μl紫杉醇溶液。阳性对照孔先加25μl浓度为10μm的维拉帕米,然后按预设的浓度加入25μl紫杉醇。继续培养48小时,每孔加25μl浓度为5mg/ml的mtt溶液,置于37℃孵育4小时。小心除去上清液,每孔加100μldmso,振荡15min使沉淀溶解。立即于酶标仪上检测吸光度(o.d.)值,检测波长为570nm。
将脱药3天生长状态良好,处于对数生长期的阿霉素耐药的悬浮细胞k562/adr以8×104个/ml的密度接种于96孔板,每孔100μl。在5%co2培养箱中37℃孵育6小时。化合物孔先加25μl浓度为10μm的化合物7a,b,然后按预设浓度加25μl紫杉醇溶液。空白对照孔先加50μl溶媒,然后按预设浓度加25μl紫杉醇溶液。阳性对照孔先加25μl浓度为10μm的维拉帕米,然后按预设的浓度加入25μl紫杉醇。继续培养48小时,每孔加25μl浓度为5mg/ml的mtt溶液,置于37℃孵育4小时。小心除去上清液,每孔加100μldmso,振荡15min使沉淀溶解。立即于酶标仪上检测吸光度(od)值,检测波长为570nm。
按下式求浓度为10μm的化合物7a,b对不同浓度阿霉素和紫杉醇对耐药细胞生长的抑制率。生长抑制率=[(对照孔的od值-化合物孔的od值)/空白孔的od值]×100%。实验重复3次,计算ic50值。表2的数据表明,化合物7a-b可有效逆转k562/adr细胞耐药且活性优于维拉帕米。表3的数据表明,化合物7a-b可有效逆转k562/tax细胞耐药且活性优于维拉帕米。这些数据表明,本发明有显著的技术效果。
表2化合物7a,b(10μm)对阿霉素抑制耐药细胞k562/adr的影响
a)与阿霉素+维拉帕米比p<0.01;n=3.
表3化合物7a,b(10μm)对阿霉素抑制耐药细胞a562/tax的影响
a)与阿霉素+维拉帕米比p<0.01;n=3.
实施例11测定化合物7a,b的抗炎活性
因为二甲苯引起的小鼠耳肿胀被公认为急性炎症模型,所以本发明在二甲苯引起的小鼠耳肿胀模型上测定化合物7a,b的治疗作用。因为阿司匹林是治疗急性炎症的阳性药,所以本发明选择阿司匹林为阳性对照药。icr雄性小鼠(体重22±3g)在22℃环境静息2天,自由饮水和进食。之后,随机分为生理盐水组(剂量为0.2ml/只),阿司匹林组(剂量为1.11mmol/kg),化合物6组(剂量为1μmol/kg)及化合物7a,b组(剂量为0.1μmol/kg),每组10只小鼠。测定时小鼠按所在组或腹腔注射生理盐水,或腹腔注射阿司匹林,或腹腔注射化合物6,或腹腔注射化合物7a,b。给药30min后,往小鼠的左耳廓均匀涂抹30μl二甲苯,2h后小鼠接受乙醚麻醉,断颈处死,剪下左右两耳,用7mm的打孔器在两耳的相同位置取圆形耳片,称重,求出两耳肿胀差值作为肿胀度。即肿胀度=左耳圆片重量–右耳圆片重量。数据见表4。在0.1μmol/kg剂量下化合物7a,b不仅有效地抑制二甲苯引起的小鼠耳肿胀,而且活性与剂量比它们大10倍的化合物6没有显著性差异。这些数据表明,本发明有显著的技术效果。
表4化合物7a,b对二甲苯引起的小鼠耳肿胀的影响
a)与生理盐水比p<0.01,与化合物6比p>0.05;n=10。
- 还没有人留言评论。精彩留言会获得点赞!