两种二核含钆磁共振成像对比剂及其制备与应用的制作方法
本发明涉及磁共振成像领域,具体涉及两种二核含钆磁共振成像对比剂及其制备与应用。
背景技术:
自1973年lauterbur首次实现磁共振成像(magneticresonanceimaging,mri)以来,磁共振成像技术以其无创、无痛苦、精确定位与近似解剖图谱般的图像显示等特点受到科学界的高度重视,并在医学领域不断扩展,已成为影像学检查中最重要的医学影像设备之一。但是磁共振成像的分辨率在某些情况下达不到临床要求,为增强成像效果需要在进行磁共振检查之前给予磁共振对比剂。据文献报道,目前临床mri中,超过35%的mri检查需要使用对比剂。其中,钆类磁共振成像对比剂临床应用最为普遍。截止目前,美国fda已经批准的钆类磁共振对比剂共有九种,可大致分为两大类,一类是以dtpa为钆离子螯合基团的链状分子,一类是以dota为钆离子螯合基团的环状分子。
目前临床使用的对比剂大部分为非特异性细胞外液间隙对比剂,它们在脑部成像效果较好,同时也可用于血管成像,但无靶向性。gd-bopta和gd-eob-dtpa因含有亲脂性的芳基基团,是两种具有肝靶向的磁共振对比剂,它们均属于以dtpa为钆离子螯合基团的链状分子。但研究显示,链状分子对比剂比环状分子对比剂的热力学稳定性和动力学稳定性差,更容易导致部分病人的肾源性系统性纤维化,也更容易在病人脑部发生钆离子沉积。
因此,开发毒性更低的环状分子对比剂以及具有器官靶向性的对比剂,是目前磁共振成像对比剂的研究热点。
技术实现要素:
本发明的目的是为了解决现有技术中的链状分子对比剂热力学稳定性和动力学稳定性相对较差,更容易导致部分病人的肾源性系统性纤维化以及在病人脑部发生钆离子沉积的问题,同时,也是为了解决现有技术中的大部分对比剂无器官靶向性的问题,从而提供两种毒性更低的具有器官靶向性尤其是肝靶向性的环状分子对比剂及其制备方法。
两种二核含钆磁共振成像对比剂,其结构式如图所示:
所述的两种二核含钆磁共振成像对比剂,其特征在于结构上含有两部分,一部分能够增强磁共振成像效果,其结构如图所示:
一部分为含苯环的亲脂性基团,其结构如图所示:
所述的两种二核含钆磁共振成像对比剂的制备方法,其特征在于该方法包括下列步骤:
其中,r=
(ⅰ)圆底烧瓶中依次加入化合物1和二甲醛2,再加入甲醇溶解,加热回流,tlc监测反应进程,反应结束后,静置冷却,过滤,蒸除溶剂得产物粗品,经重结晶得到化合物3;
(ⅱ)氮气保护下,圆底烧瓶中将化合物3溶于二氯甲烷中,再加入三氟乙酸,室温下搅拌反应48h,反应结束后,减压蒸除未反应的三氟乙酸,重结晶得产品4;
(ⅲ)圆底烧瓶中,加入化合物4并溶于纯水中,加入gdcl3水溶液,用1mnaoh调节溶液ph值,60°c反应24h,冷却后过滤,将溶液置于c-18硅胶柱,先用大量水洗,后用甲醇-水淋洗液梯度洗脱得产品溶液,冻干机冻干溶液得产品5;
所述的制备方法步骤i中化合物1和二甲醛2的摩尔比为2:1,重结晶溶剂为乙酸乙酯;
所述的制备方法步骤ii中二氯甲烷和三氟乙酸比例为1:1,重结晶溶剂为乙酸乙酯;
所述的制备方法步骤iii中gdcl3水溶液浓度为0.05m,溶液ph值为6.5~7.0;
所述的两种二核含钆磁共振成像对比剂的用途,其特征在于该两种对比剂可以用作磁共振对比剂进行体内成像;
所述的两种二核含钆磁共振成像对比剂的用途,其特征在于该两种对比剂可以用于肝靶向成像。
本发明的有益效果:
1、本发明的两种二核含钆磁共振成像对比剂分子不带电荷呈中性,为非离子型对比剂;
2、本发明的两种二核含钆磁共振成像对比剂采用大环类dota作为钆离子螯合体,提高了对比剂的热力学和动力学稳定性,可克服dtpa链状类对比剂容易导致部分病人的肾源性系统性纤维化以及更容易在病人脑部发生钆离子沉积等问题;
3、本发明的两种二核含钆磁共振成像对比剂其总弛豫效率分别达到17.3mm-1s-1和11.1mm-1s-1,远高于目前临床使用的对比剂(一般约3.5mm-1s-1),因此实际使用中可在较低浓度下得到同样的成像效果,可降低对比剂对人体的伤害,也可得到更清晰的成像图像;
4、本发明的两种二核含钆磁共振成像对比剂含有苯基取代基团,因引入了疏水基团从而增强了亲脂性能,尤其对肝脏具有选择性成像特性,可以作为肝靶向磁共振成像对比剂。
附图说明
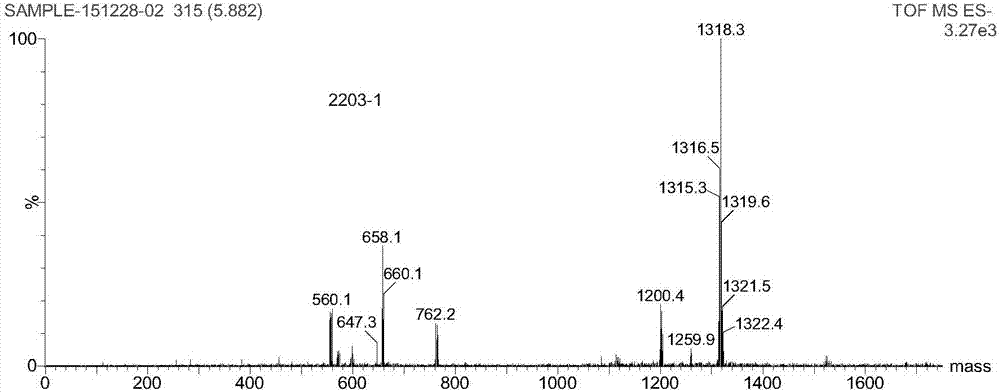
图1为对比剂a的质谱图;
图2为对比剂b的质谱图;
图3为对比剂a的弛豫效率测试图;
图4为对比剂b的弛豫效率测试图;
图5为对比剂肝脏靶向磁共振成像效果测试结果。
实施例1对比剂a的制备
(ⅰ)100ml圆底烧瓶中加入2mmol化合物1和2mmol对苯二甲醛,加入30ml甲醇溶解,加热回流,tlc监测反应进程(10%甲醇/二氯甲烷),反应结束后,静置冷却,过滤,蒸除溶剂得产物粗品,乙酸乙酯重结晶得白色固体3a,收率82%;
(ⅱ)氮气保护下,100ml圆底烧瓶中,加入1.5mmol化合物3a并溶于10ml二氯甲烷,加入10ml三氟乙酸,室温下搅拌48h,反应结束后,减压蒸除未反应的三氟乙酸,乙酸乙酯重结晶得白色固体4a,收率99%;
(ⅲ)圆底烧瓶中,加入1.0mmol化合物4a并溶于20ml纯水中,加入0.05mgdcl3水溶液22ml,用1mnaoh调节溶液ph值至6.5,60°c反应24h,冷却后过滤,将溶液置于c-18硅胶柱,先用大量水洗,后用甲醇-水淋洗液梯度洗脱得产品溶液,冻干机冻干溶液得白色固体5a(对比剂a),收率85%。
实施例2对比剂a的制备
(ⅰ)100ml圆底烧瓶中加入2mmol化合物1和2mmol对苯二甲醛,加入30ml甲醇溶解,加热回流,tlc监测反应进程(10%甲醇/二氯甲烷),反应结束后,静置冷却,过滤,蒸除溶剂得产物粗品,乙酸乙酯重结晶得白色固体3a,收率82%;
(ⅱ)氮气保护下,100ml圆底烧瓶中,加入1.5mmol化合物3a并溶于10ml二氯甲烷,加入10ml三氟乙酸,室温下搅拌48h,反应结束后,减压蒸除未反应的三氟乙酸,乙酸乙酯重结晶得白色固体4a,收率99%;
(ⅲ)圆底烧瓶中,加入1.0mmol化合物4a并溶于20ml纯水中,加入0.05mgdcl3水溶液22ml,用1mnaoh调节溶液ph值至7.0,60°c反应24h,冷却后过滤,将溶液置于c-18硅胶柱,先用大量水洗,后用甲醇-水淋洗液梯度洗脱得产品溶液,冻干机冻干溶液得白色固体5a(对比剂a),收率90%;c40h56gd2n12o14,esi-msm/z:620.7[m-2h]2-。
实施例3对比剂b的制备
(ⅰ)100ml圆底烧瓶中加入2mmol化合物1和2mmol4,4'-联苯二甲醛,加入30ml甲醇溶解,加热回流,tlc监测反应进程(10%甲醇/二氯甲烷),反应结束后,静置冷却,过滤,蒸除溶剂得产物粗品,乙酸乙酯重结晶得白色固体3b,收率85%;
(ⅱ)氮气保护下,100ml圆底烧瓶中,加入1.5mmol化合物3b并溶于10ml二氯甲烷,加入10ml三氟乙酸,室温下搅拌48h,反应结束后,减压蒸除未反应的三氟乙酸,乙酸乙酯重结晶得白色固体4b,收率100%;
(ⅲ)圆底烧瓶中,加入1.0mmol化合物4b并溶于20ml纯水中,加入0.05mgdcl3水溶液22ml,用1mnaoh调节溶液ph值至6.8,60°c反应24h,冷却后过滤,将溶液置于c-18硅胶柱,先用大量水洗,后用甲醇-水淋洗液梯度洗脱得产品溶液,冻干机冻干溶液得白色固体5b(对比剂b),收率88%;c46h60gd2n12o14,esi-msm/z:1318.3[m-h]-。
弛豫效率测试:
所有制备对比剂样品原样配制浓度为0.25mmol/l,0.5mmol/l,1.0mmol/l,2.0mmol/l,5.0mmol/l的对比剂溶液,使用核磁共振分析软件,用ir序列采集样品信号,利用反演软件反演出相应弛豫信息,ir实验参数为:p90(us)=16,p180(us)=32,td=31265,sw(khz)=100,tw(ms)=20000,rg1(db)=10,drg1=3,ns=4,nti=25。
所制备对比剂弛豫率普遍高于目前临床使用对比剂gd-dtpa(3.5mm-1s-1),该两种二磁共振对比剂总的弛豫效率分别达到17.3mm-1s-1和11.1mm-1s-1,使得实际使用中可在相对较低的浓度下获得与目前临床用对比剂同样效果,降低了对比剂使用风险,且成像效果更清晰,测试图见图3和图4。
对比剂肝靶向磁共振成像效果测试:
对比剂a、b,以gd-dota作为对照药物组,取雄性sd大鼠(280~300g),对比剂按钆浓度0.1mmol/kg尾静脉注射后再分别对大鼠肝脏进行ce-t1w扫描,扫描时间点为给药前和给药后2h;层厚3mm,间隔0.2mm,fov100×100cm2,矩阵224×192,nex2次;t1wse序列:tr550ms,te24ms,扫描后在geaw4.3工作站下处理数据,对比肝脏和正常肌肉成像效果,结果见附图5;由图可见,与gd-dota不同,对比给药前,对比剂a和b给药后2小时均能观测到肝脏部位成像明显增强,说明注射对比剂后,肝脏吸收了部分对比剂且在肝脏停留时间较长,可用于肝脏靶向成像,所制备对比剂是一类潜在的肝靶向磁共振对比剂。
具体实施方式
两种二核含钆磁共振成像对比剂,其结构式如图所示:
所述的两种二核含钆磁共振成像对比剂的制备方法,其特征在于该方法包括下列步骤:
其中,r=
(ⅰ)圆底烧瓶中依次加入化合物1和二甲醛2,再加入甲醇溶解,加热回流,tlc监测反应进程,反应结束后,静置冷却,过滤,蒸除溶剂得产物粗品,经重结晶得到化合物3;化合物1和二甲醛2的摩尔比为2:1,重结晶溶剂为乙酸乙酯;
(ⅱ)氮气保护下,圆底烧瓶中将化合物3溶于二氯甲烷中,再加入三氟乙酸,室温下搅拌反应48h,反应结束后,减压蒸除未反应的三氟乙酸,重结晶得产品4;二氯甲烷和三氟乙酸比例为1:1,重结晶溶剂为乙酸乙酯;
(ⅲ)圆底烧瓶中,加入化合物4并溶于纯水中,加入gdcl3水溶液,用1mnaoh调节溶液ph值,60°c反应24h,冷却后过滤,将溶液置于c-18硅胶柱,先用大量水洗,后用甲醇-水淋洗液梯度洗脱得产品溶液,冻干机冻干溶液得产品5;gdcl3水溶液浓度为0.05m,溶液ph值为6.5~7.0;最终产品结构经过质谱鉴定。
- 还没有人留言评论。精彩留言会获得点赞!