测序数据处理系统和SMN基因检测系统的制作方法
本发明属于基因测序技术领域,具体涉及一种测序数据处理系统和smn基因检测系统。
背景技术:
脊髓性肌萎缩症(spinalmuscularatrophy,sma)是指一组脊髓前角细胞变性所致的近端肌肉无力和萎缩的遗传性神经肌肉疾病。运动神经元生存基因(survivalmotorneuron,smn)为其致病基因,包括smn1和smn2。smn1和smn2在遗传病基因检测中一直存在着困难,主要有两个原因:一是两个致病基因在一个局部重复区域,两者在基因组中位置接近,而且序列几乎完全相同,仅存在一个可供区分两个基因的变异位点;二是这两个基因在群体中的拷贝数变异对其致病性非常重要,而且在人群中的变异数目较高。
目前,通过采用多重连接依赖式探针扩增技术(multiplexligation-dependentprobeamplification,mlpa)或定量pcr(qpcr)来检测smn1的7号外显子的缺失。这些方法的主要缺点体现在:1)提供的信息较少,实验方法比较为繁琐,很难与目前常用的检测流程直接整合;2)精度较低,无法有效检出smn2基因的拷贝数量;3)传统检测方法通常无法有效区分smn1和smn2的7号外显子的突变位点差异,并且通常也不检测其他外显子上的突变位点;4)成本较高,传统方法检测在检测其突变位点和拷贝数时,存在着实验流程繁琐、精度低、准确度差、成本高的缺点。
技术实现要素:
本发明的目的在于克服现有技术的上述不足,提供一种测序数据处理系统和smn基因检测系统,旨在解决现有smn基因检测方法的实验流程繁琐,以及精度低、准确度差的技术问题。
为实现上述发明目的,本发明采用的技术方案如下:
一方面,本发明提供一种测序数据处理系统,包括:
数据获取单元:用于获得高通量测序的含有smn基因的测序数据;
序列比对单元:用于对参考基因组中smn2基因的所有外显子注释,并将所述测序数据与注释后的所述参考基因组进行序列比对,获得所述测序数据中的匹配序列;
信息确定单元:用于根据所述匹配序列和smn基因中7号外显子的差异碱基位点,确定所述测序数据中smn基因的变异信息。
相应地,本发明上述测序数据处理系统对应一种测序数据处理方法,该测序数据处理方法包括如下步骤:
获得高通量测序的含有smn基因的测序数据;
对参考基因组中smn2基因的所有外显子注释,并将所述测序数据与注释后的所述参考基因组进行序列比对,获得所述测序数据中的匹配序列;
根据所述匹配序列和smn基因中7号外显子的差异碱基位点,确定所述测序数据中smn基因的变异信息。
另一方面,本发明提供一种smn基因检测系统,包括:
提取单元:用于提取人源离体样本中含有smn基因的核酸;
测序单元:用于对所述核酸进行高通量测序获得测序数据;
分析单元:利用本发明的测序数据处理系统分析所述测序数据,得到人源离体样本中smn基因的序列信息。
相应地,本发明上述smn基因检测系统对应一种smn基因检测方法,该smn基因检测方法包括如下步骤:
提取人源离体样本中含有smn基因的核酸;
对所述核酸进行高通量测序获得测序数据;
利用本发明的测序数据处理系统或方法分析所述测序数据,得到人源离体样本中smn基因的序列信息。
本发明提供的测序数据处理系统或方法,利用高通量测序获得的测序数据,通过生物信息学分析,不但能全面和精确检测到smn1和smn2序列,获得各种突变位点和拷贝数信息,提供更多的致病基因信息,而且还能与目前常用的检测流程直接整合,有效提高检测的易用性,降低检测的成本,有效克服了传统方法的缺点,具有速度快、准确度高的特点。
本发明提供的smn基因检测系统或方法,利用上述测序数据处理系统或方法处理高通量测序的含有smn基因的测序数据,可获得smn1和smn2的突变位点和拷贝数信息,具有速度快、准确度高、成本低的特点,为临床smn基因检测提供了一种更好的选择。
附图说明
图1为实施例2中参考基因组中smn2注释前后,smn的测序序列定位对比图;
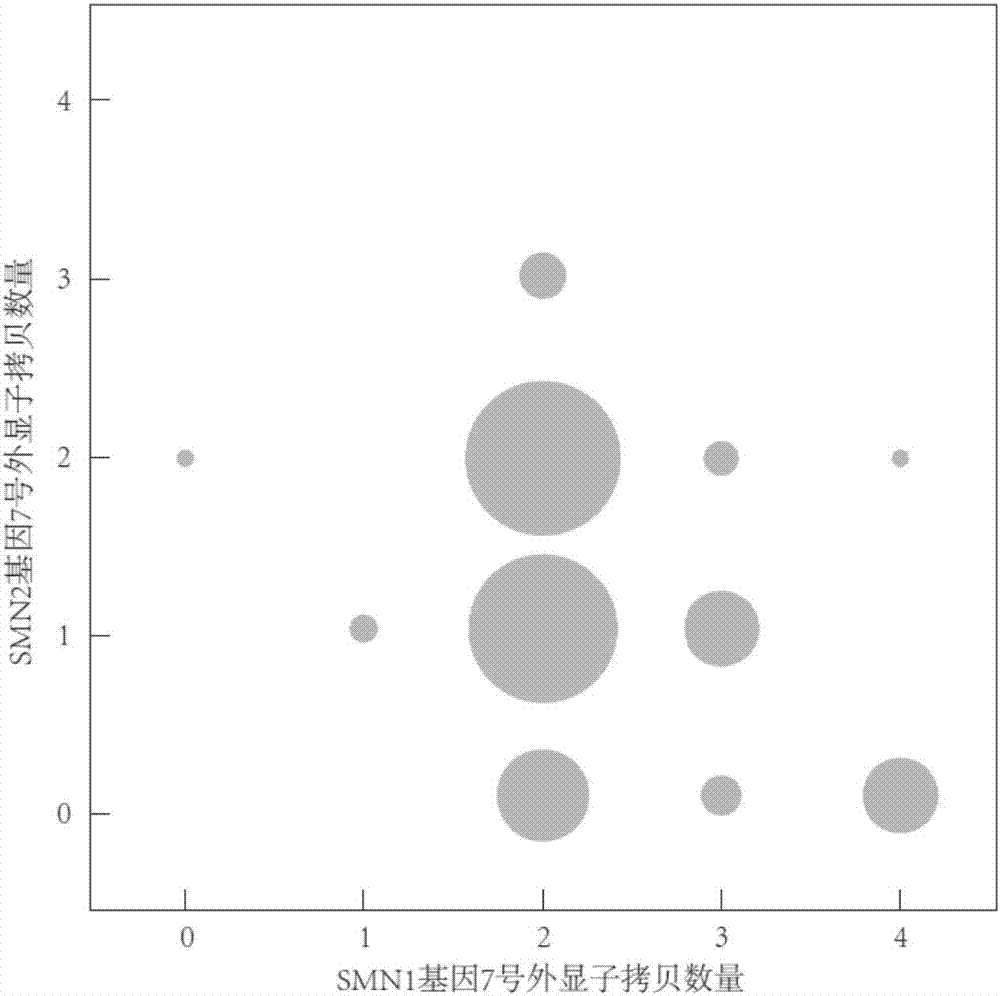
图2为实施例4的待测样本中smn1和smn2的7号外显子拷贝数结果图;
图3为实施例4的待测样本中smn1和smn2的所有外显子拷贝数结果图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合附图和实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
一方面,本发明实施例提供一种测序数据处理系统,包括
数据获取单元:用于获得高通量测序的含有smn基因的测序数据;
序列比对单元:用于对参考基因组中smn2基因的所有外显子注释,并将数据获取单元获得的测序数据与注释后的参考基因组进行序列比对,获得该测序数据中的匹配序列;
信息确定单元:用于根据序列比对单元获得的匹配序列和已知的smn基因中7号外显子的差异碱基位点,确定测序数据中smn基因的变异信息。
本实施例的上述测序数据处理系统对应一种测序数据处理方法,具体包括如下步骤:
s011:获得高通量测序的含有smn基因的测序数据(对应上述数据获取单元)。
s012:对参考基因组中smn2基因的所有外显子注释,并将上述测序数据与注释后的参考基因组进行序列比对,获得该测序数据中的匹配序列(对应上述序列比对单元)。
s013:用于上述匹配序列和已知的smn基因中7号外显子的差异碱基位点,确定测序数据中smn基因的变异信息(对应上述信息确定单元)。
目前,一般对smn基因的所有外显子定义为1-7号,而2号外显子包括外显子2a和外显子2b,因此本实施例中对smn基因所有外显子定义为:1号外显子,2a号外显子、2b号外显子、3号外显子、4号外显子、5号外显子、6号外显子、7号外显子(因7号外显子不编码蛋白质,实际上还是有七个外显子)。本实施例的测序数据处理系统或方法,在高通量测序数据的序列比对(sequencealignment)过程中,对参考基因组的smn2的1至7号外显子(第五号染色体:69344512-69373860碱基对,尽管smn2的7号外显子不编码蛋白,但是由于其与6号外显子距离很近,同样也被注释为x)被全部注释为x(即把序列变为x)。因此,在序列比对时,所有smn1和smn2的测序序列都会被定位到smn1基因上。
其中,smn1的1至7号外显子位于第五号染色体如下位置(hg19版本参考基因组):
1号外显子:70220911-702210311;
2a号外显子:70234646-70234757;
2b号外显子:70237196-70237355;
3号外显子:70238165-70238405;
4号外显子:70238525-70238717;
5号外显子:70240465-70240600;
6号外显子:70241873-70242023;
7号外显子70247748-70247838。
本发明一实施例中,将参考基因组smn2注释为x,通过计算机模拟,结果显示本实施例的方法能将smn1和smn2两个基因的测序序列都准确定位到smn1上。
该测序数据处理系统或方法,将参考基因组smn2注释为x,在序列比对时,所有smn1和smn2的测序序列都会被定位到smn1基因上,然后分析7号外显子上smn1/smn2差异碱基的拷贝数量和其分布,可以判断smn1和smn2其他外显子各自的拷贝数量和变异的情况。
具体地,在上述序列比对单元或步骤s012中,序列比对可利用hwa-mem或bowtie等软件完成,本发明一实施例中,使用bwa-mem软件将测序序列匹配到注释后的参考基因组上,获得匹配序列,为后续的分析处理提供重要保证。
具体地,在上述信息确定单元或步骤s013中的分析处理过程为:从匹配序列中找到注释后的参考基因组中smn1基因上的所有突变,并结合已知的差异碱基位点(即smn1/smn2差异位点,位于五号染色体70247773位置,其中smn1是c,而smn2是t),确定测序数据中smn基因的所有突变位点,且利用隐马氏法得到smn基因的总拷贝数。该隐马氏法的公式如下:
基因组被分为m区间,c1…cm代表第1至m区间的拷贝数,oi为第i区间的序列数量。
进一步地,上述分析处理过程还包括:根据该总拷贝数和差异碱基位点,利用贝叶斯法、隐马氏法和t检验法中的任意一种得到总拷贝数中smn1基因和smn2基因的各自拷贝数。该总拷贝数可以为smn基因的任一外显子拷贝数,由于7号外显子的拷贝数对临床指导至关重要,本发明一优选实施例中,总拷贝数为7号外显子总拷贝数,并利用了贝叶斯法计算得到smn1基因和smn2基因的7号外显子的各自拷贝数,具体过程为:
首先获得smn1和smn2的7号外显子的总的拷贝数量n,再通过7号外显子的差异,采用贝叶斯方法估测smn1和smn27号外显子各自的拷贝数量n1,n2,公式如下:
nb指负二项分布,
该算法原理明确,考虑了突变和拷贝数变异检测中两个致病基因序列容易混淆的问题,避免了过去方法中流程繁琐、准确度差的问题。通过一个整合的算法,可以高效、精确获得smn1和smn2的突变和拷贝数变异情况。
另一方面,本发明实施例提供一种smn基因检测系统,包括:
提取单元:用于提取人源离体样本中含有smn基因的核酸;
测序单元:用于对提取单元获得的核酸进行高通量测序获得测序数据;
分析单元:利用本实施例的测序数据处理系统或方法分析从测序单元中得到的测序数据,从而得到人源离体样本中smn基因的序列信息。
相应地,本发明实施例的上述smn基因检测系统对应一种smn基因检测方法,该smn基因检测方法包括如下步骤:
s021:提取人源离体样本中含有smn基因的核酸;
s022:对上述核酸进行高通量测序获得测序数据;
s023:利用本实施例测序数据处理系统或方法分析上述测序数据,得到人源离体样本中smn基因的序列信息。
本发明提供的smn基因检测系统或方法中,人源离体样本包括血液、尿液和唾液中的至少一种,高通量测序的测序平台为illumina测序平台或iontorrent测序平台,如illumina公司的hiseq-2500、hiseq-2000等。利用本实施例的测序数据处理系统或方法对通过这些测序平台获得的测序数据进行分析处理,可获得人源离体样本样本中smn1和smn2的突变位点和拷贝数信息,该smn基因检测方法可作为一种离体非诊断性检查方法,具有速度快、准确度高、成本低的特点。
本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
实施例1
一种测序数据处理方法,包括如下步骤:
s111:获得高通量测序的含有smn基因的测序数据。
s112:对参考基因组中smn2基因的所有外显子(第五号染色体:69344512-69373860碱基对,1至7号外显子)全部注释为x,并利用bwa-mem软件将测序数据与注释后的参考基因组进行序列对比,获得测序数据中的匹配序列。
s113:从匹配序列中找到注释后的参考基因组中smn1基因上的所有突变,并结合差异碱基位点(即smn1/smn2差异位点,位于五号染色体70247773位置,其中smn1是c,而smn2是t),确定测序数据中smn基因的所有突变位点,且利用隐马氏法得到smn基因的7号外显子的总拷贝数,隐马氏法公式如下:
基因组被分为m区间,c1…cm代表第1至m区间的拷贝数,oi为第i区间的序列数量。
然后,再通过7号外显子的差异,采用贝叶斯方法估测smn1和smn2的7号外显子各自的拷贝数量n1,n2,贝叶斯方法公式如下:
nb指负二项分布,
实施例2
计算机模拟测试实施例1中参考基因组的注释定位效果:
通过将参考基因组中的smn2外显子序列注释为x,smn1和smn2两个基因的测序序列都准确定位到了smn1上,定位结果如图1所示:图1中的第一行是smn1的1-7号外显子,第二行是smn2的1-7号外显子;其中空心箱线图表示用标准的参考基因组(未注释)进行基因定位,记为原参考基因组(p),而深色实心的箱线图表示将参考基因组smn2用x注释后的基因定位,记为注释后参考基因组(m),横坐标表示四个不同的测试数据集(具体为:sr1:48个样本;sr2:48个样本;sr3:48个样本;sr4:48个样本),纵坐标表示唯一定位到的测序序列数量。
由图1中结果可知,在原有的标准参考基因组的分析中,smn1和smn2在1-6号外显子上定位到的测序序列都很少,而本实施例中将参考基因组smn2用x注释后,在smn1的1-6号外显子上定位到的测序序列数量明显更多了,而由于7号染色体上70247724和70247773位置的snp,p和m两种方式定位到的测序序列都比较好,而m方法仍然优于p方法。
实施例3
参考基因组注释后的对照组(不含smn区域的测序数据)和实验组(含有smn区域的测序数据)的测序序列匹配情况进行对比,详细分析结果如表1和表2所示。
表1为对照组:dna捕获不含smn区域(即不含smn1和smn2区域);表2为实验组:dna捕获含smn区域(即含smn1和smn2区域)。从下述表1和表2的数据结果表明:在参考基因组注释后,之前无法唯一匹配测序序列成功匹配到smn1上,之前匹配到smn2的测序序列也匹配到smn1上,而对基因组的其他区域影响不大。
表1
表2
实施例4
使用119个样本获得了smn1编码区的突变,利用注释参考基因组进行数据处理获得的突变信息如下表3,获得拷贝数变异情况如图2和图3所示(图中圆圈的大小,代表样本数量的多少)。
表3
从图2可知,smn1和smn2基因的7号外显子拷贝数都很高,约40%的测试样本内smn2基因的7号外显子拷贝数是1个。从图3可知:对smn1和smn2基因,其1-6号外显子的拷贝数总量高于两者7号外显子的拷贝数总量。以上数据结果表明,本实施例的测序数据处理系统或方法在不影响全基因组其他基因注释和分析的前提下,可以准确有效地检测出smn1和smn2各自的突变和拷贝数变异。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!