降低CRISPR/Cas9介导的胚胎基因编辑脱靶率的方法与流程
本发明涉及基因编辑,具体涉及如何降低crispr/cas9介导的胚胎基因编辑的脱靶率。
背景技术:
伴随遗传学和基因组学新时代的到来,先进的dna测序技术发现了调控个体发育异常的分子密码。基因编辑,曾经被称为基因打靶技术,主要依赖特异的碱基互补配对原则修改基因组dna序列。目前,大多数的基因组编辑借助自定义的内切酶得以实现,包括锌指结构核酸酶(zfns),转录激活样效应核酸酶(talens)以及crispr-cas9系统。zfns及talens技术需要复杂的蛋白质装配工程,而crispr-cas9系统可以通过替换引导rna轻松地完成。因此,作为一个高效,设计简便,使用方便,多重靶向编辑的基因编辑工具,crispr-cas9系统被广泛应用于各项研究,成为当前主流的基因编辑工具,为实现精准的基因编辑带来希望。
crispr/cas9系统目前已应用于体外的人诱导多能干细胞及初始t细胞的基因编辑研究中,我们也进一步证明基因编辑可以增强t细胞的功能[suetal.,2016]。然而对于很多遗传疾病的研究,例如肌强直性营养不良,遗传性线粒体疾病等,仅限于对体细胞的基因编辑的研究,并不能达到预防疾病的目的,因而,对生殖细胞的基因编辑的研究,可以有助于进一步理解不孕症及早期胚胎发育中的一些关键科学问题。
在人类胚胎研究中,已证实受精卵中显微注射zfn,talen及cas9等核酸酶,可以实现多个物种二倍体胚胎中dna、mrna或蛋白水平的基因编辑。目前仅在异常受精的、不能发育为存活胚胎的三原核受精卵中做了有关cas9的研究,但发现crispr/cas9系统存在效率及准确率受限的不足,尤其是在研究中频繁出现的脱靶现象。虽有多种方法尝试改善crispr/cas9基因编辑技术高频的脱靶现象,但仍不能避免其带来的各种突变缺陷。
因而进一步降低crispr/cas9介导的对人二倍体胚胎基因编辑的脱靶效应尤为迫切。
技术实现要素:
本发明的目的在于提供一种降低crispr/cas9介导的胚胎基因编辑脱靶率的方法。
本发明基于二倍体胚胎能完全排除三原核受精卵带来的未知因素干扰,为检测基因靶向效应提供良好的研究模型,通过在卵子受精瞬间同时导入cas9mrna以及sgrna来实现基因编辑。
根据本发明的降低crispr/cas9介导的胚胎基因编辑脱靶率的方法包括:
合成靶向基因的sgrna序列,并构建携带t7启动子的sgrna表达质粒;
体外转录cas9mrna及sgrna;以及
采取卵胞浆内单精子显微注射技术(icsi)将单精子导入未受精的mii期卵子,并同时导入cas9mrna及sgrna。
本发明的方法还可以包括:对所得受精卵进行胚胎培养和评估。
根据本发明的优选实施例,所述cas9mrna为cas9切口酶mrna,所述sgrna为两个相邻位置的sgrna。这种情况下,所述sgrna进一步优选为sgrna-e和sgrna-1,它们距离rag1基因编码位点11bp。
本发明利用ivf过程中无法正常受精的废弃卵子,通过同时导入精子、cas9mrna以及sgrna(优选cas9mrna和双sgrna)使其受精形成二倍体胚胎,并进行了crispr/cas9介导的基因编辑测试。通过深度测序及t7en1裂解实验详细分析crispr/cas9在人类二倍体胚胎的基因编辑的特异性,可以看出,使用本发明方法的靶向编辑效率很高,脱靶突变率很低,通过采取cas9切口酶/双sgrna策略能够成功地降低脱靶突变率。此外,crispr/cas9系统还能诱导多重基因靶向,证明其在人类胚胎的基因编辑中用途广泛、容易操控。
附图说明
图1是荧光原位杂交方法检测icsi注射后受精并可发育的二倍体胚胎。
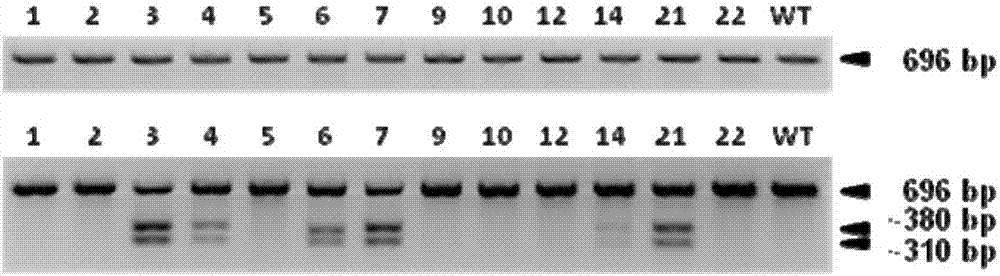
图2是通过t7en1裂解实验在废弃卵子来源的胚胎中检测cas9介导的靶向rag1基因裂解,其中,所用sgrna为rag1-sgrna-e。从人胚胎基因组dna中经pcr扩增得到rag1基因位点的靶向区域。pcr的产物(上)经过t7en1裂解分析(下)。wt,野生对照。
图3是通过t7en1裂解实验在废弃卵子来源的胚胎中检测cas9介导的靶向rag1基因裂解,其中,所用sgrna为rag1-sgrna-1。从人胚胎基因组dna中经pcr扩增得到rag1基因位点的靶向区域。pcr的产物(上)经过t7en1裂解分析(下)。pcr扩增所用的引物显示在表1中。
图4是废弃卵子来源的胚胎中修饰编辑后的rag1基因序列。pam序列用下划线标记,靶向序列用淡色标记,突变序列用淡色、小写字母标记;缺失(-),插入(+)。n/n代表测序过程中的阳性比例。
图5是rag1-sgrna-e的可能脱靶位点的pcr产物分析。从3、4、6、7、14和21号胚胎扩增与rag1-sgrna-e最为同源的25个可能脱靶位点。在pp13的7号中观察到了突变带。wt,野生对照。
图6是通过t7en1裂解实验分析rag1-sgrna-e可能的脱靶位点。t7en1裂解实验检测从3、4、6、7、14和21号胚胎扩增出与rag1-sgrna-e最为同源的25个可能脱靶位点。其中7号胚胎潜在的pp13位点的突变通过深度测序确认。wt,野生对照。
图7是rag1-sgrna-1的可能脱靶位点的pcr产物分析。从68、70、72、78、80和82号胚胎扩增与rag1-sgrna-1最为同源的12个可能脱靶位点。均未观察到突变带。wt,野生对照。
图8是通过t7en1裂解实验分析rag1-sgrna-1可能的脱靶位点。t7en1裂解实验检测从68、70、72、78、80和82号胚胎扩增出与rag1-sgrna-1最为同源的12个可能脱靶位点。72、80和82号潜在的pp29位点的突变未通过深度测序确认。wt,野生对照。
图9是cas9/sgrna介导废弃卵子来源的胚胎中基因多位点编辑,废弃卵子来源的胚胎中rag1基因靶向序列的pcr产物。从胚胎基因组dna中pcr扩增rag1的靶向序列。pcr扩增使用的引物显示在表1中。wt,野生型。
图10是cas9/sgrna介导废弃卵子来源的胚胎中基因多位点编辑,通过t7en1裂解实验验证cas9/sgrna介导的rag1基因不同序列的靶向裂解。对图9中获得的pcr产物进行t7en1裂解分析。
图11是cas9/sgrna介导废弃卵子来源的胚胎中基因多位点编辑,废弃卵子来源的胚胎中rag1等位基因修饰编码后的序列。pam序列用下划线标记;靶向序列用淡色标记;突变序列用淡色、小写字母标记;缺失(-),插入(+)。n/n代表测序过程中的阳性比例。
图12是cas9切口酶/双sgrna介导废弃卵子来源的胚胎中基因编辑,废弃卵子来源的胚胎中rag1基因靶向序列的pcr产物。从胚胎基因组dna中pcr扩增rag1的靶向序列。pcr扩增使用的引物显示在表1中。wt,野生型。
图13是cas9切口酶/双sgrna介导废弃卵子来源的胚胎中基因编辑,通过t7en1裂解实验验证cas9/sgrna介导的rag1基因不同序列的靶向裂解。对图11中获得的pcr产物进行t7en1裂解分析。
图14是cas9切口酶/双sgrna介导废弃卵子来源的胚胎中基因编辑,废弃卵子来源的胚胎中rag1等位基因修饰编码后的序列。pam序列用下划线标记;靶向序列用淡色标记;突变序列用淡色、小写字母标记;缺失(-),插入(+)。n/n代表测序过程中的阳性比例。
图15是cas9/sgrna介导的废弃卵子来源的胚胎中多基因编辑,检测cas9/sgrna介导的靶向rag1基因裂解。从人胚胎基因组dna中经pcr扩增得到rag1基因位点的靶向区域(上)。pcr的产物经过t7en1裂解分析(下)。pcr扩增所用的引物显示在表1中。wt,野生型对照。
图16是cas9/sgrna介导的废弃卵子来源的胚胎中多基因编辑,检测cas9/sgrna介导的靶向ube3a基因裂解。从人胚胎基因组dna中经pcr扩增得到ube3a基因位点的靶向区域(上)。pcr的产物经过t7en1裂解分析(下)。
图17是cas9/sgrna介导的废弃卵子来源的胚胎中多基因编辑,废弃卵子来源的胚胎中修饰编辑后的rag1和ube3a基因序列。pam序列用下划线标记,靶向序列用淡色标记,突变序列用淡色、小写字母标记;缺失(-),插入(+)。n/n代表测序过程中的阳性比例。
具体实施方式
实施例1
本实施例1中,选择了常规ivf中不能正常受精的废弃卵子,通过icsi方法促进这些卵子受精并发育成二倍体胚胎,我们在废弃卵子胞浆内单精子注射的同时采用联合注射已证实可以靶向人类编码重组激活基因1(rag1)的有效sgrna-e与cas9mrna。随后,采用t7en1裂解方法及深度测序检测cas9/sgrna技术在人二倍体胚胎基因编辑靶向效果和脱靶效率。
1.1使用本发明提供的方法,利用crispr/cas9系统对废弃卵子来源的人二倍体胚胎进行基因编辑:
1)废弃的人类卵子的来源:
所有不孕症患者夫妇均签署在体外受精-胚胎移植周期中因卵子未受精形成废弃卵子和授精后多余精子形成废弃精子捐献用于科学研究的知情同意书。废弃配子与精子用于科学研究分别于2006年5月11日(2006002)及2008年4月11日(2008001)通过南京大学医学院附属鼓楼医院伦理委员会的审核。自2014年4月至2014年10月,共计66枚未正常受精的mii卵子纳入此研究,所有卵子均由南京大学医学院附属鼓楼医院生殖中心体外受精(ivf)周期中通过促排卵方案获得。
2)质粒构建:
合成含c端nls的cas9/cas9d10a人源密码子优化的序列并克隆至pst1374载体的t7启动子之后,通过ncbi的blast同源性分析rag1基因序列,选择避免大量相似位点的5‘-n(20)ngg-3’基序的序列,合成的sgrna的寡核苷酸经退火后克隆至bsai线性化的puc57-t7sgrna载体以构建携带t7启动子的sgrna表达质粒。
表1:用于pcr扩增sgrna靶向基因的引物信息
3)cas9mrna及sgrna的体外转录:
cas9/cas9d10amrna及sgrnas的体外转录操作各步骤均按相应试剂盒所述方法操作。简述如下:pst1374-cas9/cas9d10a-nls质粒经plasmidmidi试剂盒(qiagen,12143)提取,agei-hf(neb,r3552)消化,pcr纯化试剂盒(qiagen,28004)纯化后用mmessagemmachinet7ultrakit(ambion,lifetechnologies,am1345)体外转录获得cas9mrna。cas9mrna采用rneasyminikit(qiagen,74104)纯化。puc57-t7sgrna质粒经axyprepplasmidminiprep试剂盒(axygen,ap-mn-p-250g)提取,drai(neb,r0129)消化,pcr纯化试剂盒(qiagen,28004)纯化,megashortscriptkit试剂盒(ambion,lifetechnologies,am1354)等所述方法进行体外转录,并采用megaclearkit(ambion,lifetechnologies,am1908)所述方法进行纯化sgrna。
4)胞浆内注射(icsi):
卵胞浆内单精子显微注射按palermo等所述方法进行,简述如下:将未受精的mii期卵子移入icsi专用的培养皿中从第一极体的6点及12点钟方向固定卵子以确保注射针头与卵泡膜在同一水平线上。将吸有单一精子的注射针小心插入卵子透明带并轻柔地突破卵泡膜后将精子注射到卵子的细胞质中。每个废卵注射精子同时注射2.5pl含20ng/μlcas9mrna和10ng/μlsgrnas的混合rnas。5)胚胎培养及评估:
将注射后的卵子单独置于g1培养基(vitrolife,sweden)中培养,注射后16-18小时评估是否受精。分别于精子注射后43-45小时及67-69小时对卵裂期胚胎的形态,包括卵裂球数量,对称性和碎片的程度等进行评估。胚胎评估标准采用2012年伊斯坦布尔共识。注射后72小时收集所有胚胎并冻存于-80℃备用。
1.2t7en1裂解实验检测基因编辑靶向效果:
其中一些可以受精并且发育至二倍体胚胎(图1)(表2),部分注射精子的卵子可以受精,并发育至2-11细胞阶段。在最初的两次检测中,分别收集了10个(#1-10)与12个(#11-22)胚胎(表2),并对每个胚胎的全基因组进行了扩增以检测靶向效果。
具体方法如下:
1.2.1单个胚胎全基因组扩增:
单个胚胎采用repli-gsinglecellkit试剂盒(qiagen,150345)所述方法进行胚胎全基因组扩增。dna产物用无dna酶和rna酶的水进行1:10至1:100稀释进行下一轮pcr扩增。
1.2.2t7en1裂解实验:
以稀释的dna为模板,使用primestarhsdnapolymerase(takara,dr010)将rag1,ube3a的靶向序列框及可能的脱靶位点进行pcr扩增,并且使用pcrclean-upkit(axygen,ap-pcr-250)纯化。纯化的pcr产物(200ng)在nebuffer2(neb)中逐渐降温完成变性及退火。在37℃水浴中用0.3μl的t7en1(neb,m0302)消化杂交pcr产物25分钟,再用2.5%琼脂糖胶电泳分离。
其结果显示,分别有9个(#1-7,9-10)及4个(#12,14,21-22)胚胎的基因组可以正确扩增(比率分别是9/10,4/12)。
表2:显微注射cas9mrnaandsgrnas的胚胎结果统计
利用这些全基因组扩增所得的dna,我们对每个样本的靶向序列连同附近的序列均通过pcr进行扩增,随后进行t7en1裂解实验,结果显示:4个(#3,4,6,7)以及2个(#14,21)扩增产物出现了明显的裂解条带(图2&表2),提示这些胚胎成功完成了基因编辑。
随后,我们进一步用不同的sgrna证明cas9对人胚胎基因编辑的靶向效率。我们换用另外一种已经证实有效靶向rag1基因的sgrna-1进行了3次重复实验。分别收集3个(#68-70),7个(#71-77,),6个(#78-83)样品,均完成了基因组dna扩增。靶向分析结果显示,三次实验中分别有2个(#68,70),1个(#72),2个(#78,80)胚胎出现裂解条带(图3&表2)。
此外,通过基因测序及pcr产物的ta克隆测序进一步确认上述基因编辑的靶向效果(图4&表3)。pcr扩增所用的引物显示在表1中。值得注意的是,对于sgrna-e,靶向效率由部分胚胎(#1-2,5,9-10,12,22)的无法检测到个别胚胎的100%(#3)差异很大。结果表明,cas9系统可以实现及其高效的基因编辑。
表3:sanger测序结果统计分析结果
荧光原位杂交方法(图1使用的方法以检测注射胚胎染色体完整性)
单一精子注射72小时后,透明带打孔后固定胚胎,使透明带缺口位于2点钟处,用平口活检针从缺口处轻轻吸出一枚卵裂球进行荧光原位杂交检测染色体完整性。卵裂球经固定液(0.01nhcl/0.1%tween20)处理溶解细胞膜及细胞质,暴露细胞核并固定至玻片上。空气干燥后,用2×枸橼酸钠溶液(ssc,promega,catalogue#v4261)室温清洗5分钟,然后分别经过70%,85%及100%乙醇梯度脱水。
荧光原位杂交通过14(spectrumred,catalogue#kbi-40226r)及21号(spectrumgreen,catalogue#kbi-40238g)染色体端粒近端探针进行杂交。所有卵裂球样本和探针采用thermobritehybridization系统(model:s500-24)中进行73℃变性3分钟,37℃杂交孵育16小时。杂交完成后,50%甲酰胺/2×ssc以及2×ssc43℃分别漂洗玻片三次。0.5μg/ml的4,6-diamidino-2-phenylindole(dapiii,vysis,catalogue#32-804831)复染后,奥林巴斯bx51荧光显微镜观察染色情况。使用appliedimaging系统及相应2.8版本软件进行图像采集与分析。
1.3脱靶分析和对脱靶候选序列的深度测序:
对所有成功靶向基因编辑的二倍体胚胎样本进行了脱靶效应的检测。为了查找其潜在的脱靶位点,实验涉及的sgrna-e及sgrna-1均通过一个开放的搜索软件seqmap(http://www‐personal.umich.edu/~jianghui/seqmap/)与全基因组进行了同源性分析。sgrna-e及sgrna-1分别存在25个及12个可能的脱靶位点见表4。在脱靶分析中,采用为检索确定寡核苷酸对应基因组信息而设计的seqmap软件进行脱靶分析。在检索过程中,靶向序列错配参数被设置为5。‘ngg’被选作pam。最终,保留了最接近pam的7bp并且总错配数不超过4个的脱靶序列作为t7en1裂解实验与测序的候选。
表4:候选的脱靶位点
深度测序适用于小样本量的分析,其检测脱靶效应灵敏度最高,因此我们首先用其检测经不同引物对扩增的所有预测位点(表4&5)。对脱靶候选序列的深度测序时,用表5中的引物对脱靶序列进行pcr扩增,pcr产物使用pcrclean-upkit(axygen,ap-pcr-250)纯化。利用纯化的pcr产物构建4个dna文库,使用truseqnanokit试剂盒,包括1个dna文库qc(20%phixcontrollibrary),用miseq(pe2*250)测序,标记出脱靶位点附近出现单核苷酸多态性及插入缺失。
表5:用于pcr检测rag1‐sgran‐e和rag1‐sgran‐1潜在37个脱靶位点的pcr扩增引物序列
结果显示,仅#7样品在ots13上检测到一段60bp的缺失,说明cas9/sgrna诱导的脱靶突变发生率极低(1/37)。为了进一步确认深度测序检测到的脱靶突变结果,我们采取了更适合个体样本脱靶现象分析的t7en1裂解实验(图5-8),结果证实仅#7样品在ots13处发现了裂解带(图6)。同样地,采用桑格测序进行验证也发现了一个60bp的缺失(表3)。这些结果表明,在人类二倍体胚胎中采用crispr/cas9进行基因编辑诱导的脱靶突变率极低。
cas9/sgrna系统在2细胞胚胎阶段前完成了所有的基因靶向,所有检测的胚胎中结果显示少于4种基因型(表3)。靶向效率从0%到100%差异很大,说明获得令人满意的靶向效率是可行的。
实施例2
使用cas9切口酶/双sgrna策略减少脱靶损伤
在本实施例中,尝试将需要同时应用2个相邻位置的sgrna的编辑策略应用于人二倍体胚胎。我们选择了sgrna-e和sgrna-1,它们相距11bp。两次独立实验分别收集了4个(#84-87)和3个(#95-97)人二倍体胚胎。全基因组dna经过pcr扩增后用于t7en1酶切分析,在5个样本中发现了酶切条带(#84-87,#95)(图9&10和表2)。这一结果与既往研究一致:双sgrna策略较单sgrna策略有着更高的靶向效率(11/29vs5/7)(表2)。pcr产物直接测序结果显示ta克隆产物有着多个峰值进一步证实了这一结果(图11&表3)。同样,靶向效率从无法检测的0%至100%(#84)也说明cas9系统可以有效的工作。
在证明了cas9和双sgrna的复合靶向作用后,检测了cas9切口酶/双sgrna策略的效果。选择使用d10acas9切口酶,在两次独立实验中分别收集了5个(#107-111)和4个(#112-115)胚胎。除了108号胚胎未能扩增基因组dna,其余胚胎的基因组dna都经成功扩增后用于t7en1酶切分析。结果显示:一半的样本(#110-113)存在酶切条带(图12&13),这说明cas9切口酶/双sgrna在人类胚胎中成功诱导了基因靶向编辑。所有的裂解结果都经过pcr产物测序进一步验证,产物中ta克隆和之前描述的一样有着多个峰值(图14&表3)。有趣的是其中一个样本(#111)的pcr产物中只有一个截短了25bp大小的单峰(图13&表3),说明此胚胎中发生了纯合的基因靶向修饰。
我们进一步开始检测该系统是否能降低脱靶率。检测靶向编辑成功的样品(#110-113)后并未在37个预测的ots中发现裂解条带,这其中包括在野生型cas9酶中产生脱靶裂解条带的ots13。进一步通过在野生型cas9检测的样本(#3,4,6,7,14,21)和cas9d10a检测的样本(#110-113)中同时重复ots13的检测确认,仅在同一个野生型的cas9检测样本(#7)中发现了裂解条带和截短了60bp的条带,证明cas9切口酶/双sgrna系统能有效降低人类胚胎中脱靶率。
在随后应用的双sgrna策略中,我们实现了靶向效率的进一步提高,并且得到了双位点突变的胚胎(表3)。特别是通过双sgrna策略,我们完成了几个纯合的基因编辑(#111&147)(图13&16,表3)。这些都说明之前所担心的人类胚胎中靶向效率低的问题是可以通过优化crispr/cas9系统和胚胎操作加以提高。
实施例3
crispr/cas9系统能介导人类胚胎中复合的基因组工程
为了验证crispr/cas9系统是否能介导人类胚胎中多基因编辑,我们设计了两个靶向ube3a基因(编码泛素蛋白连接酶e3a)的sgrna,即ube3a-sgrna-1和2,连同rag1-sgrna-e和1同时微注射到人类胚胎中。两次独立实验中分别收集了4个(#147-150)和6个(#151-156)胚胎。第一次实验中的4个胚胎都顺利完成全基因组扩增,第二次实验中的6个胚胎里有4个顺利完成了全基因组扩增,扩增后的dna进行t7en1酶切实验。结果显示(图15&16):8个胚胎中,5个(#147-150,152)存在rag1靶向的酶切条带,3个(#149-150,152)存在ube3a靶向的酶切条带。值得我们注意的是,8个胚胎中有3个(#149-150,152)同时存在rag1和ube3a的突变(图15&16),说明在人类胚胎中可以实现多基因编辑。pcr产物的测序结果中出现的ta克隆多峰值进一步证明了这种突变体的大小和靶向效率(图17和表3)。上述结果表明在不同的胚胎中存在不同效率的rag1和ube3a插入缺失(图17和表3)。有趣的是,有2个胚胎中不存在野生的rag1基因。其中一个(#147)只有rag1突变,另一个同时有突变的rag1和ube3a(图17和表3),这说明crispr/cas9甚至可以诱导纯合的复合基因修饰。
- 还没有人留言评论。精彩留言会获得点赞!