Rakicidin衍生物,制备方法及其在制备抗癌药物中的用途与流程
本发明提供了一种rakicidin衍生物,并涉及rakicidin衍生物的制备方法,及其在制备治疗癌症药物中的用途,本发明属于药物技术领域。
背景技术:
rakicidin是由小单胞菌no.r385-2a(单胞菌属)产生的,具有抗癌活性的一系列化合物的总称。rakicidin系列化合物是一种包含15元环的环状脂肽类化合物。
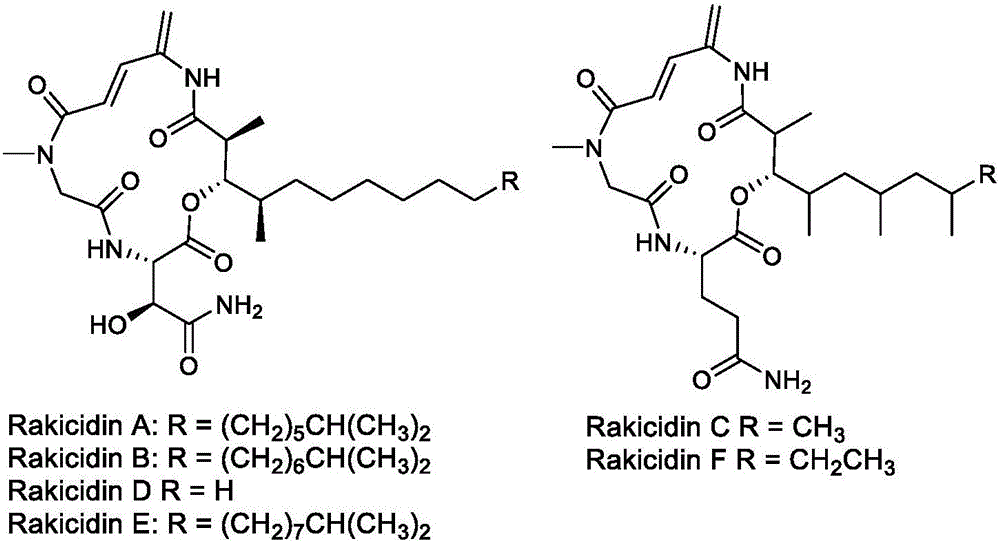
根据r基团的差别将化合物命名为rakicidina、rakicidinb、rakicidinc、rakicidind、rakicidine和rakicidinf六个天然产物,其结构见说明书附图1。报道称,be-43547系列化合物对k562、k562/g+、ba/f3cellsexpressingbcr-ablt315i和panc-1等癌细胞有抑制活性。2016年,poulsen等人探究了rakicidina对胰腺癌细胞panc-1的细胞活性,其中,在正常氧含量条件下ic50为400nm,而在缺氧条件下的ic50提高到37nm。以rakicidina为例,rakicidina对胰腺癌细胞panc-1在缺氧下的细胞毒性是正常氧条件下的11倍。而缺氧是许多实体瘤的基本特征,并且与肿瘤的转移和化疗耐受相关。因此,研究像rakicidina这样的,具有低氧选择细胞毒性的化合物,对于癌症的治疗研究是非常重要的。rakicidins系列化合物与抗菌素vinylamycin和具有抗癌活性化合物be-43547在结构上有类似性。这些化合物都是大环酯肽类天然产物,都含有一种非天然氨基酸4-氨基-2,4-戊二烯酰胺结构片段。此类化合物在抗肿瘤方面均表现出了较好的生物活性,有进一步研究开发的潜力。但是,此类化合物的全合成路线长、总产率低及稳定性较差。对于此类化合物的简化结构修饰,可能有利于增强其成药性。本发明通过对β-羟基天冬氨酸片段和侧链的修饰,合成了rakicidin衍生物,该衍生物具有治疗癌症的作用。
技术实现要素:
本发明提供了一种如式(i)所示的rakicidn衍生物,
式(i)中r1为氢、烷基、环烷基、羟基取代烷基、烯基、炔基、芳基、烷基芳基、芳基烷基、芳基烯基、芳基炔基、杂环基、三氟甲基、多氟取代烷基、腈基、腈基甲基、酰基、氨基甲酰基、磺酰基、磺酰胺基或芳氧烷基;r2为异丙基或正丙基。
一种制备式(i)所示rakicidin衍生物的方法,其特征在于以式(ii)所示的化合物为原料制备式(i)所示的化合物,
一种如式(i)所示的rakicidin衍生物,在制备治疗癌症或治疗癌症的辅助药物中的用途,其中癌症为白血病、乳腺癌、前列腺癌、鼻咽癌、大肠癌、肺癌、肝癌、食道癌、胃癌、肠道癌、肾癌、口腔癌、何杰金淋巴癌、胰腺癌、直肠结肠癌、子宫颈癌、非何杰金淋巴癌、神经胶质瘤、脑瘤、黑瘤、膀胱癌、卵巢癌、甲状腺癌或卡波西肉瘤。
一种用于治疗癌症的药物组合物,其中含有有效量的式(i)rakicidin衍生物在药学上可接受的载体或与其他抗癌药物的组合物。
附图说明
图1.天然产物rakicidin的结构通式
图2.化合物4和11的合成
图3.化合物17的合成
具体实施方式
为了理解本发明,下面以实施例进一步说明本发明,但不意于限制本发明的保护范围。
实施例1:rakicidin衍生物及其盐的合成
具体合成路线见说明书附图2和说明书附图3,具体步骤如下:
氩气保护下,向化合物8(35.7g,70.0mmol)的四氢呋喃(350ml)溶液依次加入四三苯基磷钯(16.19g,14mmol)和n-甲基苯胺(15g,140mmol),于室温下搅拌反应1.5h,并用乙酸乙酯(500ml)稀释反应液。有机相用1%hcl(120ml)洗涤两次。得到的有机相用无水硫酸钠干燥,过滤后浓缩,得到的粗品用硅胶柱纯化,得到无色油状化合物2(30.25g,92%)
250ml圆底烧瓶中,依次加入上一步得到化合物2(19.2g,40.88mmol)、肌氨酸烯丙酯(7.92g,61.32mmol)和二氯甲烷160ml。将hobt(8.29g,61.32mmol)和edci(15.67g,81.76mmol)依次加至反应体系,继续搅拌反应12h。加入10ml水至反应体系淬灭反应,然后用600ml乙酸乙酯稀释反应液后,分别用1%盐酸水溶液(300ml)、饱和碳酸氢钠(300ml)和饱和食盐水(300ml)各洗一次,将无水硫酸钠加入到盛有有机相的锥形瓶中,充分晃动混匀,静置干燥后过滤,浓缩。得到的粗品用硅胶柱纯化,得到的产物3为无色油状物(22.0g,93%)。[α]24d=+72.0(c=0.5,chcl3);1hnmr(400mhz,dmso-d6)δ7.59(s,4h),7.47–7.32(m,6h),7.20–7.02(m,1h),6.73–6.34(m,2h),5.93–5.78(m,1h),5.28(d,j=17.3hz,1h),5.18(d,j=10.5hz,1h),4.57(s,2h),4.44–4.20(m,2h),4.15(s,1h),3.64–3.49(m,2h),3.07(s,2h),2.85(s,1h),1.36(s,9h),0.98(d,j=10.5hz,9h);13cnmr(101mhz,dmso-d6)δ169.1,169.0,166.0,165.7,155.1,143.1,142.3,135.1,132.9,132.3,132.2,129.9,127.9,121.1,120.9,118.0,117.8,79.2,78.0,65.6,65.4,65.1,64.8,53.5,50.7,49.3,36.2,34.4,28.1,26.6,18.8;ir(neat)νmax:3308,2931,2858,1747,1710,1665,1623,1168,1109,702,504cm-1;hrms(esi)calcdforc32h44n2nao6si+[m+na]+:603.2866,found603.2865.
氩气保护下,将化合物5(300.3g,690mmol)和hmpa(240.8g,1360mmol)的四氢呋喃(1500ml)溶液置于-78℃下搅拌。向反应液中加入nahmds的四氢呋喃溶液(430.0ml,860.0mmol,2mol/l的四氢呋喃溶液)。搅拌30分钟后,将碘甲烷(240.77g,1700mmol)缓慢滴加至反应液。继续搅拌反应3小时后,加入饱和氯化铵水溶液(600ml)萃灭反应。将反应体系升至室温,萃取分液,得到的有机相用无水硫酸钠干燥,浓缩,得到的粗品用硅胶柱纯化,得到的产物6为白色固体物(280.4g,90%)。[α]23d=-58.4(c=1.5,chcl3);1hnmr(400mhz,cdcl3)δ3.88(t,j=6.3hz,1h),3.45(q,j=13.8hz,2h),3.08–2.97(m,1h),2.03(d,j=6.3hz,2h),1.96–1.81(m,3h),1.80–1.69(m,1h),1.45–1.20(m,28h),1.17(d,j=6.9hz,3h),1.14(s,3h),0.95(s,3h),0.88–0.83(m,3h);13cnmr(100mhz,cdcl3)δ176.5,65.2,53.3,48.3,47.8,44.7,40.5,38.6,32.9,32.8,32.0,29.8,29.8,29.7,29.6,29.5,27.4,26.5,22.8,20.9,20.0,19.1,14.2;ir(neat)νmax:2920,2849,1679,1460,1328,1249,1165,1134,1114,722,681,534,512cm-1;hrms(maldi)calcdforc27h49nnao3s+[m+na]+:490.3325,found490.3328.
氩气保护下,将化合物6(80.3g,177.0mmol)的二氯甲烷(800ml)溶液置于-78℃下搅拌。向反应液中加入1mol/l的dibal-h的正己烷溶液(220ml,220mmol)。搅拌3小时后,依次加入h2o(8.8ml),15%naoh(8.8ml)和h2o(26.4ml)淬灭以上反应,反应液用正己烷(100ml)稀释,水相用正己烷(50ml)萃取两次,合并有机相用无水硫酸钠干燥,浓缩,得到的粗品用硅胶柱纯化,得到的产物7为无色油状物直接用于下一步反应。
氩气保护下,向化合物8(60.22g,221.1mmol)的二氯甲烷(300ml)溶液依次加入et3n(31.44g,310.76mmol)和tbsotf(78.88g,298.18mmol),于室温下搅拌反应过夜。旋转蒸发除去可挥发性试剂,向得到的残渣里加入正戊烷(800ml),搅拌10分钟。将上层的正戊烷层缓慢倾倒出后,旋蒸浓缩得到的粗品9溶于二氯甲烷(300ml),直接用于下一步反应。
氩气保护下,将化合物7的二氯甲烷(100ml)溶液置于-78℃下搅拌。向反应液中加入三氟化硼乙醚溶液(15.13g,105.17mmol)。搅拌30分钟后,将上一步得到的化合物9的二氯甲烷溶液滴加至反应液。继续搅拌反应3h后,加入饱和氯化铵水溶液(250ml)萃灭反应。将反应体系升至室温,萃取分液,得到的有机相用无水硫酸钠干燥,浓缩,得到粗品化合物10,直接用于下一步反应。
氩气保护下,将上一步得到的醇10(120.78g,269mmol)溶解于四氢呋喃/甲醇/水(180ml/60ml/60ml)置于室温下搅拌。向反应液中加入氢氧化锂一水合物(19.55g,67.3mmol),室温下搅拌反应4h。浓缩反应除去四氢呋喃和甲醇,将反应体系用10%nahso4酸化到ph=3,用乙酸乙酯(300ml)萃取两次,将无水硫酸钠加入到盛有合并的有机相的锥形瓶中,充分晃动混匀,静置干燥后过滤,浓缩,得到的粗品直接用于下一步反应。
500ml圆底烧瓶中,依次加入上一步得到的粗品化合物溶于dmf(250ml)。将碳酸钾(71.35g,510mmol)和烯丙基溴(50ml,590mmol)依次加至反应体系,继续搅拌反应10h。加入100ml水至反应体系淬灭反应,然后用150ml乙酸乙酯萃取反应液后,用饱和食盐水(100ml)洗涤两次,将无水硫酸钠加入到盛有有机相的锥形瓶中,充分晃动混匀,静置干燥后过滤,浓缩。得到的粗品用硅胶柱纯化,得到的产物11为无色油状物(47.59g,四步产率48%)。[α]20d=-38.6(c=1.0,chcl3);1hnmr(400mhz,cdcl3)δ5.97–5.84(m,1h),5.31(d,j=17.2hz,1h),5.22(d,j=10.4hz,1h),4.60(d,j=5.5hz,2h),3.59(s,1h),2.65(p,j=7.3hz,1h),2.48(d,j=5.7hz,1h),1.60–1.50(m,1h),1.45–1.36(m,1h),1.29–1.21(m,26h),1.15(d,j=7.4hz,3h),0.90–0.82(m,6h);13cnmr(100mhz,cdcl3)δ176.3,132.1,118.5,76.1,65.3,43.3,35.1,34.0,32.0,30.0,29.8,29.5,27.4,26.1,22.8,14.6,14.2,12.9;ir(neat)νmax:3529,2922,2853,1720,1459,1167,983,538cm-1;hrms(esi)calcdforc23h44nao3+[m+na]+:391.3183,found391.3188.
25ml圆底烧瓶中,加入化合物11(1.1g,3.0mmol)和化合物12a(1.85g,7.5mmol),并用6ml二氯甲烷溶解,于0℃搅拌。将dmap(110mg,0.90mmol)和dic(1.14g,9.00mmol)依次加至反应烧瓶中,反应体系于室温继续搅拌1.5h。然后向反应液缓慢加入1ml水淬灭反应,然后将反应液浓缩,残渣用100ml乙酸乙酯和20ml的1%的硫酸氢钠水溶液稀释后,分液。有机相用饱和碳酸氢钠(20ml)洗一次,无水硫酸钠干燥,过滤,使用旋转蒸发仪将溶液浓缩,硅胶柱层析纯化,得无色油状化合物13a(1.4g,78%)。[α]23d=-45.8(c=2.3,chcl3);1hnmr(400mhz,cdcl3)δ6.00–5.74(m,2h),5.31(d,j=17.7hz,1h),5.23(d,j=10.1hz,1h),5.15–5.04(m,1h),4.55(s,2h),4.47–4.37(m,1h),2.88–2.79(m,1h),2.75(d,j=3.2hz,3h),2.69–2.57(m,1h),1.78–1.65(m,1h),1.40(s,9h),1.22(br,26h),1.13(d,j=6.5hz,3h),0.90–0.81(m,6h);13cnmr(101mhz,cdcl3)δ174.2,171.0,170.5,155.7,132.1,118.7,79.8,78.8,65.6,50.8,41.9,37.3,34.4,33.3,32.0,29.8,29.8,29.5,28.4,27.2,26.3,22.8,14.2,14.2,13.7;ir(neat)νmax:3307,2923,2853,1738,1717,1650,1250,1164,557cm-1;hrms(maldi)calcdforc33h60n2nao7+[m+na]+:619.4293,found619.4298.
化合物13b(产率:75%)的制备方法同化合物13a。[α]23d=-43.5(c=1.2,chcl3);1hnmr(400mhz,cdcl3)δ6.01–5.69(m,2h),5.30(d,j=17.5hz,1h),5.23(d,j=10.3hz,1h),5.16–5.01(m,1h),4.55(s,2h),4.39(d,j=7.5hz,1h),2.90–2.76(m,1h),2.70–2.47(m,2h),1.79–1.65(m,1h),1.40(s,9h),1.22(brs,26h),1.14(d,j=7.1hz,3h),0.89–0.81(m,6h),0.73–0.67(m,2h),0.52–0.44(m,2h);13cnmr(100mhz,cdcl3)δ174.2,171.3,170.9,155.7,132.1,118.7,79.8,78.7,65.7,50.7,41.9,37.3,34.4,33.3,32.0,29.8,29.8,29.5,28.4,27.2,22.8,22.7,14.3,14.2,13.7,6.4,6.2;ir(neat)νmax:3350,2923,2853,1738,1717,1651,1249,1165,555cm-1;hrms(maldi)calcdforc35h62n2nao7+[m+na]+:645.4449,found645.4452.
氩气保护下,向化合物13a(1.25g,2.10mmol)的四氢呋喃(21ml)溶液依次加入四三苯基磷钯(485mg,0.42mmol)和n-甲基苯胺(450mg,4.2mmol),于室温下搅拌反应1.5h,并用乙酸乙酯(50ml)稀释反应液。有机相用1%hcl(120ml)洗涤两次。得到的有机相用无水硫酸钠干燥,过滤后浓缩,得到的粗品用硅胶柱纯化,得到相应是酸直接用于下一步反应。
250ml圆底烧瓶中,加入化合物3(26g,44.8mmol)并用200ml二氯甲烷溶解,于0℃下搅拌。将三氟乙酸(22ml)加至反应体系,反应体系继续搅拌30min后,转移至室温反应6h。将反应液缓慢加入500ml饱和碳酸氢钠溶液中,然后用乙酸乙酯(300ml)萃取3次,将无水硫酸钠加入到盛有有机相的锥形瓶中,充分晃动混匀,静置干燥后过滤,浓缩,得到黄色油状化合物4,不需纯化直接用于下一步反应。
250ml圆底烧瓶中,依次加入上一步得到的酸、化合物4(1.51g,3.15mmol)和二氯甲烷8.4ml。将hobt(426mg,3.15mmol)和edci(805mg,4.2mmol)依次加至反应体系,继续搅拌反应12h。加入10ml水至反应体系淬灭反应,然后用200ml乙酸乙酯稀释反应液后,分别用1%盐酸水溶液(100ml)、饱和碳酸氢钠(100ml)和饱和食盐水(100ml)各洗一次,将无水硫酸钠加入到盛有有机相的锥形瓶中,充分晃动混匀,静置干燥后过滤,浓缩。得到的粗品用硅胶柱纯化,得到的产物14a为无色油状物(1.39g,两步产率65%)。
化合物14b(产率:53%)的制备方法同化合物14a。
氩气保护下,向化合物14a(1.39g,1.37mmol)的四氢呋喃(13.7ml)溶液依次加入四三苯基磷钯(317mg,0.27mmol)和n-甲基苯胺(294mg,2.75mmol),于室温下搅拌反应1.5h,并用乙酸乙酯(40ml)稀释反应液。有机相用1%hcl(120ml)洗涤两次。得到的有机相用无水硫酸钠干燥,过滤后浓缩,得到的粗品用硅胶柱纯化,得到相应是酸直接用于下一步反应。
25ml圆底烧瓶中,加入上述化合物并用9ml二氯甲烷溶解,于0℃下搅拌。将三氟乙酸(2.3ml)加至反应体系,反应体系继续搅拌30min后,转移至室温反应2h。向反应液加入5ml甲苯稀释反应液,然后将反应液旋蒸除去有机溶剂,得到的残渣为白色固体,不需纯化直接用于下一步反应。
2l圆底烧瓶中,加入hatu(6.50g,17.10mmol)和1100ml无水thf,于室温下搅拌,然后将dipea(5.97ml,34.2mmol)加入。将上一步得到的白色固体溶解于20ml无水thf中,使用注射泵将溶液缓慢加至反应液中,滴加过程超过8h。滴加完毕,继续搅拌12h后,将反应液旋干,得到棕黄色残渣。将残渣用300ml乙酸乙酯:甲醇=2:1的混合溶剂溶解,硅藻土过滤,滤液使用旋转蒸发仪将溶液浓缩后,溶解于250ml乙酸乙酯中。然后分别用1%的盐酸水溶液(60ml)、饱和碳酸氢钠水溶液和饱(60ml)和食盐水(60ml)洗涤。有机相用无水硫酸钠干燥,过滤,浓缩,硅胶柱层析纯化,得到白色固体化合物15a(685mg,三步产率58%)。
[α]22d=-139.6(c=0.3,dmso);1hnmr(400mhz,dmso-d6)δ8.31(d,j=8.9hz,1h),7.92(d,j=8.8hz,1h),7.90–7.85(m,1h),7.61(t,j=6.7hz,4h),7.48–7.38(m,6h),6.69(dd,j=14.9,2.6hz,1h),6.01(d,j=14.8hz,1h),5.14(d,j=10.4hz,1h),4.74–4.58(m,2h),4.34(d,j=18.1hz,1h),3.78(d,j=18.1hz,1h),3.65–3.59(m,1h),3.58–3.49(m,1h),2.93(s,3h),2.83–2.76(m,1h),2.59–2.51(m,4h),2.43(dd,j=15.1,4.4hz,1h),1.73–1.62(m,1h),1.22(br,26h),1.02–0.92(m,15h),0.84(t,j=6.6hz,3h);13cnmr(100mhz,dmso-d6)δ173.0,169.7,168.5,167.5,165.5,142.0,135.1,135.1,132.8,132.7,130.0,129.9,127.9,119.3,76.9,65.2,52.1,51.1,49.3,41.6,38.4,36.4,33.6,33.2,31.3,29.2,29.0,28.7,27.0,26.6,25.5,22.1,18.8,15.8,13.9,13.1;ir(neat)νmax:3280,2923,2853,1740,1663,1611,1551,1278,1111,701,504cm-1;hrms(maldi)calcdforc49h76n4nao7si+[m+na]+:883.5375,found883.5378.
化合物15b(三步产率:52%)的制备方法同化合物15a。
[α]22d=-145.1(c=0.5,dmso);1hnmr(400mhz,dmso-d6)δ8.40(d,j=9.1hz,1h),8.05(d,j=4.1hz,1h),7.98(d,j=8.9hz,1h),7.60(t,j=6.7hz,4h),7.48–7.34(m,6h),6.67(dd,j=15.0,2.6hz,1h),6.03(d,j=14.0hz,1h),5.13(d,j=10.4hz,1h),4.74–4.56(m,2h),4.37(d,j=18.1hz,1h),3.73(d,j=18.0hz,1h),3.63–3.57(m,1h),3.56–3.48(m,1h),2.92(s,3h),2.87–2.76(m,1h),2.61–2.53(m,1h),2.37(dd,j=15.0,4.4hz,1h),1.71–1.61(m,1h),1.21(br,26h),1.00–0.90(m,15h),0.82(t,j=6.6hz,3h),0.59–0.52(m,2h),0.37–0.31(m,2h);13cnmr(100mhz,dmso-d6)δ173.0,169.7,169.1,167.5,165.5,142.0,135.1,135.0,132.8,132.7,129.9,129.9,127.9,119.3,76.8,65.2,54.9,52.1,51.2,49.3,41.6,38.5,36.4,33.6,33.3,31.3,29.2,29.1,28.7,27.0,26.5,22.2,22.1,18.8,15.8,13.9,13.1,5.6,5.4;ir(neat)νmax:3284,2923,2854,1740,1665,1609,1551,1280,1112,701,504cm-1;hrms(maldi)calcdforc51h78n4nao7si+[m+na]+:909.5532,found909.5535.
10ml圆底烧瓶中,将化合物15a(189mg,0.22mmol)溶解于thf(3ml)中,依次加入hoac(38μl,0.65mmol)和tbaf(0.65ml,0.65mmol,1mthf溶液),继续搅拌反应12h。加入60ml乙酸乙酯和20ml水稀释反应液,水相用乙酸乙酯(3×30ml)萃取。合并的有机相用无水硫酸钠干燥,过滤后浓缩。得到的粗品用硅胶柱纯化,得到化合物16a。
10ml圆底烧瓶中,加入化合物16a(84mg,0.135mmol)并用2.7ml无水thf溶解,于0℃下搅拌。将三乙胺(47μl,0.338mmol)和乙基磺酰氯(25μl,0.27mmol)依次加至反应体系,反应体系继续搅拌30min后。向反应液加入200μl水,使用旋转蒸发仪将溶液浓缩,残渣用etoac(80ml)溶解,无水硫酸钠干燥,浓缩得到残渣,未经纯化直接用于下一步反应。
10ml圆底烧瓶中,加入上一步得到的白色固体并溶解于5.4ml无水thf中,与室温下搅拌。然后加入dbu(0.64g,1.08mmol),继续搅拌反应1h。然后加入乙酸乙酯稀释后,用1%硫酸氢钠水溶液(20ml)、饱和碳酸氢钠水溶液(10ml)和饱和食盐水(10ml)各洗涤一次,无水硫酸钠干燥,使用旋转蒸发仪将溶液浓缩至大致为3ml的浓缩液,将浓缩液用硅胶柱纯化,得到的溶液使用旋转蒸发仪将溶液浓缩至大致为3ml的浓缩液,加入3ml叔丁醇稀释,继续使用旋转蒸发仪将溶液浓缩至大致为3ml的浓缩液,再加入3ml蒸馏水,用冷冻干燥机干燥,得到粉末状化合物17a(65mg,三步产率49%)。
[α]18d=-48.6(c=0.2,meoh/chcl3=4:1);1hnmr(400mhz,meod-d4/cdcl3=4:1)δ7.16(d,j=15.0hz,1h),6.30(d,j=15.1hz,1h),5.62(s,1h),5.57(s,1h),5.35(d,j=9.5hz,1h),4.99–4.93(m,1h),4.54(d,j=17.7hz,1h),3.97(d,j=17.8hz,1h),3.22(s,3h),3.07–2.96(m,1h),2.87(dd,j=15.6,6.4hz,1h),2.78(s,3h),2.74(dd,j=15.6,4.3hz,1h),1.99–1.84(m,1h),1.36(br,26h),1.30(d,j=6.9hz,3h),1.12(d,j=6.7hz,3h),0.97(t,j=6.6hz,3h);13cnmr(100mhz,meod-d4/cdcl3=4:1)δ172.5,169.4,169.2,167.6,165.9,138.5,138.1,118.7,116.5,77.5,52.4,49.3,41.9,38.4,36.5,33.8,33.1,31.3,31.1,29.8,29.2,29.1,29.0,28.7,26.9,22.1,22.1,15.2,13.9,13.1,5.6,5.4;ir(neat)νmax:3251,2920,2851,1743,1679,1639,1611,1544,1259,1238,990,693,530cm-1;hrms(maldi)calcdforc33h56n4nao6+[m+na]+:627.4092,found627.4098.
化合物17b(三步产率:43%)的制备方法同化合物16a。[α]18d=-41.5(c=0.1,dmso/chcl3=3:1);1hnmr(400mhz,dmso-d6/cdcl3=3:1)δ8.83(s,1h),8.21(d,j=8.2hz,1h),7.92(d,j=4.0hz,1h),6.84(d,j=15.0hz,1h),6.15(d,j=15.0hz,1h),5.35(s,1h),5.32(s,1h),5.04(d,j=9.8hz,1h),4.69–4.60(m,1h),4.35(d,j=17.8hz,1h),3.72(d,j=17.8hz,1h),2.96(s,3h),2.90–2.80(m,1h),2.57–2.51(m,1h),2.50–2.35(m,2h),1.72–1.62(m,1h),1.19(br,26h),1.02(d,j=6.9hz,3h),0.92(d,j=6.7hz,3h),0.81(t,j=6.7hz,3h),0.57–0.50(m,2h),0.36–0.28(m,2h);13cnmr(100mhz,dmso-d6/cdcl3=3:1)δ172.5,169.4,169.2,167.6,165.9,138.5,138.1,118.7,116.5,77.5,52.4,49.3,41.9,38.4,36.5,33.8,33.1,31.3,31.1,29.8,29.2,29.0,29.0,28.7,26.9,22.1,22.1,15.2,13.9,13.1,5.6,5.4;ir(neat)νmax:3305,2921,2852,1716,1688,1650,1614,1537,1279,1252,897,548cm-1;hrms(maldi)calcdforc35h58n4nao6+[m+na]+:653.4249,found653.4250.
实施例2:rakicidin衍生物对人胰腺癌细胞系panc-1,aspc-1和patu8988的常氧和低氧生物活性
将待测试细胞配成2×105/ml细胞悬液,加入96孔板圆底细胞培养板内,分别加入待测化合物,每一测试浓度3孔,置37℃、5%co2饱和湿度条件下培养72小时,用mtt法在酶联检测仪570nm波长测得吸光度(a)值,计算出本发明化合物对测试癌细胞的抑制作用。
表1.rakicidin衍生物对各种胰腺癌癌细胞的抑制活性(ic50,μm)
如表1所示,所测试化合物对测试的癌细胞系显示出较强的抗癌活性。
- 还没有人留言评论。精彩留言会获得点赞!