由动量传递截面分布确定分子和分子组合体结构的制作方法
本申请要求2016年9月21日提交的美国临时申请第62/397,665号的优先权,其内容通过引用整体并入本文。
本公开大体涉及由离子淌度-质谱(im-ms)数据确定分子和分子组合体结构。
背景技术:
蛋白质结构和蛋白质组合体在疾病病因学中起着至关重要的作用,因为蛋白质功能与其结构密切相关。例如,常见的神经退行性疾病帕金森氏症和阿尔茨海默氏症是由特定蛋白质的错误折叠和聚集引起的。与这些疾病相关的蛋白质类似于朊病毒样病原体,因为它们通过动态过程形成自繁殖的蛋白质组合体:单个蛋白质组合成小蛋白质组合体,并且小蛋白质组合体组合成大蛋白质组合体。这一动态过程包括解折叠和/或重排蛋白质和/或蛋白质组合体结构。更为普遍的,美国许多增长最快的重大疾病,如阿尔茨海默氏病、帕金森氏病、2型糖尿病、动脉粥样硬化和癌症,都涉及到蛋白质及其组合体的瞬时聚集构象。
核磁共振(nmr)和x射线光谱学提供了蛋白质的整体平均结构,对于药物研究来说非常有价值。然而,这些技术不太适合阐明动态蛋白质及其复合物的详细结构。现有技术不能捕捉共存的、瞬时聚集的蛋白质构象,因为它们只测量蛋白质的整体平均结构。因为用于药物开发的明确分子靶标无法识别,所以无法阐明共存的、瞬时蛋白质构象,而这阻碍了开发治疗或预防许多疾病的药理学策略。
离子淌度谱-质谱(ims-ms)已经被用于通常小的有机和无机分子的结构表征。该领域的最新进展使设备得到改进,从而使得ims-ms能够用于涉及大的大分子有机和生物化合物的研究。ims-ms能够揭示瞬时蛋白质结构,非常适合研究蛋白质及其复合物共存的、瞬时构象,因为它物理上分离了在毫秒内具有不同质量和形状的分析物。由于ms分析的高灵敏度和速度,ims-ms可以在几秒钟内使用微量样品完成。然而,ims-ms仅测量蛋白质或蛋白质组合体的取向平均截面。ims-ms数据并未揭示结构细节,仅从ims-ms数据中提取详细的分子结构就具有很大挑战性。
尚无已知的方法用于仅从测量截面中重新提取详细分子结构。现有技术的方法将来自传统技术(例如nmr)的结构与ims-ms数据分析相结合。然而,这种方法不能充分利用ims-ms的潜力,ims-ms是用来阐明无法使用传统方法的那些系统的结构。当理论(平均)模型结构的计算截面与实验数据相匹配或者使用实验截面作为″过滤器″从计算模型结构库中选择特定结构时,其他方法报告了结构;然而,这些方法有许多缺点。首先,不同的蛋白质结构可以有相同的截面。其次,实验截面可以是实验中相互转换的不同结构的截面的平均值。再次,ims-ms实验中的蛋白质动力学取决于电荷态。因此,需要能够从ims-ms数据中提取分子详细结构信息的改进系统和方法。
技术实现要素:
提供了用于确定分子的分子结构的方法。在实施例中,该方法包括:基于分子的路易斯结构,确定分子的多个候选分子结构;确定每个候选分子结构的预测时间分辨离子淌度质谱(trim-ms)光谱;至少部分地基于分子的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子结构的子集;确定子集中的每个候选分子结构的候选分数;以及基于候选分数分配分子的分子结构。
提供了用于确定分子组合体的分子结构的方法。在实施例中,该方法包括:确定分子的多个候选分子组合体;确定每个候选分子组合体的预测时间分辨离子淌度质谱(trim-ms)光谱;至少部分地基于分子的分子组合体的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子组合体的子集;确定子集中每个候选分子组合体的候选分数;以及基于候选分数分配分子的分子组合体。
提供了用于确定分子的分子结构的设备。在实施例中,该设备包括存储器和处理电路,被配置为:基于分子的路易斯结构,确定分子的多个候选分子结构;确定每个候选分子结构的预测时间分辨离子淌度质谱(trim-ms)光谱;至少部分地基于分子的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子结构的子集;确定子集中的每个候选分子结构的候选分数;以及基于候选分数分配分子的分子结构。
提供了用于确定分子组合体的分子结构的设备。在实施例中,该设备包括存储器和处理电路,被配置为:确定分子的多个候选分子组合体;确定每个候选分子组合体的预测时间分辨离子淌度质谱(trim-ms)为分子;至少部分地基于分子的分子组合体的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子组合体的子集;确定子集中每个候选分子组合体的候选分数;以及基于候选分数分配分子的分子组合体。
提供了一种存储用于确定分子的分子结构的计算机可执行指令的非暂时性计算机可读介质。在实施例中,该非暂时性计算机可读介质存储计算机可执行指令,当由一个或多个处理器执行时,所述指令执行以下操作:基于分子的路易斯结构,确定分子的多个候选分子结构;确定每个候选分子结构的预测时间分辨离子淌度质谱(trim-ms)光谱;至少部分地基于分子的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子结构的子集;确定子集中的每个候选分子结构的候选分数;以及基于候选分数分配分子的分子结构。
提供了一种存储用于确定分子组合体的分子结构的计算机可执行指令的非暂时性计算机可读介质。在实施例中,该非暂时性计算机可读介质存储计算机可执行指令,当由一个或多个处理器执行时,所述指令执行以下操作:确定分子的多个候选分子组合体;确定每个候选分子组合体的预测时间分辨离子淌度质谱(trim-ms)光谱;至少部分地基于分子的分子组合体的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子组合体的子集;确定子集中每个候选分子组合体的候选分数;以及基于候选分数分配分子的分子组合体。
附图说明
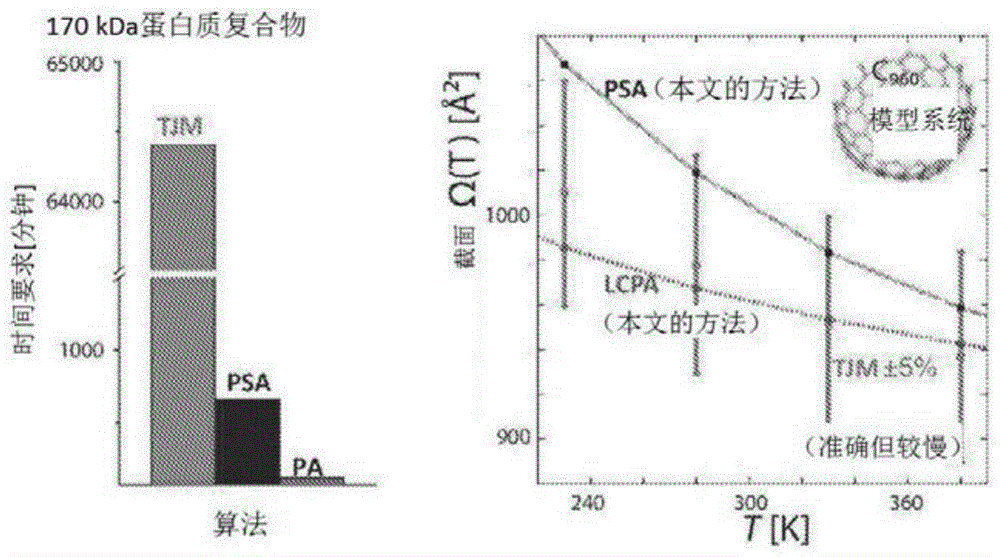
图1示出了使用局部碰撞概率逼近(lcpa)、投影逼近(psa)和轨迹法(tjm)算法的ims-ms数据的截面计算。psa和lcpa算法与tjm一样精确,但速度要快得多。投影逼近不精确(约30%误差)。
图2示出了适于根据本公开的实施例使用的ims-ms仪器。
图3a-3d显示了各种实验结果。图3a示出了时间分辨ims-ms方法的发展,包括将测量的到达时间与电动力学场中离子的截面相关联的方程。图3b示出了基于图3a中的方程,可以直接根据实验数据测量绝对截面而无需校准,并且在传统漂移管系统的误差范围内。图3c示出了时间分辨ims-ms实现了大于250的分辨率,而其他ims-ms系统实现的的分辨率在40和1oo之间。图3d示出了图2的ims设备中时间分辨测量的特征性能。
图4示出了趋化因子ccl5(6+电荷态)的测量和预测ims-ms光谱。本公开的系统和方法阐明了低聚趋化细胞因子(趋化因子)如何控制免疫细胞迁移的基本方面。
图5示出了各种电荷态下泛素的截面,其中ims-ms数据(三角形)再现了bowers数据(圆形),并且其中低电荷态的截面与nmr结构一致。
图6a-6b示出了蛋白质泛素的实验结果。图6a示出了泛素6+和7+电荷态的时间分辨ims-ms截面分布。通过″软″和碰撞激活仪器设置获得的实验数据显示了类天然特征(~nmr),这些特征在几秒钟的时间标度上是稳定的,并且出现了气相结构(见12.387秒后的重叠)。图6b将实验(6+,″软″设置,0.002秒和12.387秒,见图6a)与预测时间分辨ims-ms光谱进行了比较。宽峰表明存在几个未分辨的蛋白质构象。
图7示出了泛素6+的一种拟议的解折叠机制,部分由ims-ms数据确定。当两个中心β链之间的相互作用消失时,解折叠就开始了,这是由作为时间的函数的天然接触所揭示的。
图8示出了由仅考虑分子(例如蛋白质)的路易斯结构(例如氨基酸序列)的时间分辨ims-ms光谱预测分子的三维结构的流程图。
图9示出了预测和测量的时间分辨ims光谱(泛素、电荷态6+、7+、8+)。在″软″设置下,在o.002秒存在类天然峰值。实验和预测的时间分辨ims-ms光谱非常一致,表明本公开的系统和方法可以定量和明确地解释分子(例如蛋白质)的ims-ms光谱。
图10示出了由nmr测定的泛素的结构与根据本公开的实施例,对电荷态6+、7+、8+的致密峰
图11示出了im-ms揭示ad中包含的aβ肽的朊病毒样组合体。结构细节(例如亚基是如何结合的)不适合使用常规技术进行研究。本系统和方法能够鉴定肽如何从单个单体组合成淀粉样纤维,这是ad发病ad的关键步骤。这尤其为ad候选药物提供了分子机制。
图12示出了根据本公开的实施例的利用ims-ms确定分子组合体(例如蛋白质组合体)的详细结构的工作流程。
图13示出了通过搜索方法由im-ms揭示的蛋白质结构与nmr结构相匹配。
图14示出了根据本公开的实施例的用于确定分子的分子结构的方法。
图15示出了根据本公开的实施例的用于确定分子组合体的分子结构的方法。
图16示出了根据本公开的实施例的示例设备或系统的框图。
具体实施方式
一般描述
本公开实现了仅从ims-ms数据重新确定分子结构或组合体,包括生物相关的蛋白质结构或组合体。在较高水平上,该系统和方法:基于分子的路易斯结构对其进行全面的构象分析;通过考虑气相中电荷态特定分子动力学来预测ims-ms光谱;以及选择和报告最能解释实验光谱的结构。更具体地,该系统和方法:使用分子的路易斯结构来计算分子的多个潜在构象结构(例如3-d结构);计算潜在构象结构的预期ims-ms数据;将计算的潜在构象结构的ims-ms数据与测量的(即真实的)分子的ims-ms数据进行比较;以及至少部分地基于比较计算的ims-ms数据和测量的ims-ms数据,确定分子的分子结构。
本公开实现了在高通量自动化物质中阐明分子和分子聚集体的三维结构(例如分子的二级结构、三级结构、四级结构和聚集体结构),包括分子如蛋白质的瞬时聚集构象。本公开的一个优点是不需要分子三维结构的先验知识来确定分子的结构。该系统和方法可以将(1)分子的路易斯结构(例如蛋白质初级结构或氨基酸序列);和(2)分子的实验ims-ms光谱(例如,在实验室实验中测量的光谱)作为输入。根据这两个输入,系统和方法可以报告分子最可能的结构构象。
在一些方面,本公开的系统和方法可以鉴定分子及其复合物的共存、瞬时、相互转化的构象,这是现有系统和方法无法做到的。推导出的结构可以用于多种用途,如改进药物开发和促进疾病发现。例如,本公开实现了快速揭示复杂生物过程的分子机制,以及通过对相关分子结构特征(例如结合位点、与其他已知分子的相似性、分子如何相互连接等)的详细了解来改进药物筛选。
在示例性实施例中,本公开的系统和方法部分地通过使用计算lcpa(局部碰撞概率逼近)和psa(投影逼近)算法来计算分子的电荷态特定模型结构的预测截面,并将时间分辨的、捕获的、电荷态特定实验ims-ms光谱与预测ims-ms光谱进行比较来确定分子结构。本公开的系统和方法可以预测分子(例如蛋白质)的每个不同电荷态的时间分辨ims-ms光谱,用于与实验ims-ms光谱进行比较。psa和lcpa方法可以精确计算数千个截面,用于预测分子结构的整个ims-ms光谱。当采用所有商用ims-ms系统均使用的氮气缓冲气体进行实验时,这一点尤其适用。在实施例中,通过快速经验方法筛选可能的分子构象,并且只有短分子动力学模拟用于构象细化。
在本公开的各个部分中,参考示例性实施例描述了本发明:阐明蛋白质和蛋白质组合体结构。然而,这并非意在限制,本发明通常适用于任何有机或无机分子。因此,本文中关于确定蛋白质结构/组合体的任何示例性公开可以更普遍地应用于任何分子(例如糖、核酸等),并且确定任何类型分子的分子结构/组合体都在本发明的范围内。
本文提供了用于确定分子1400的分子结构的方法,如图14所示。该方法可以包括以下一项或多项:基于分子的路易斯结构,确定分子的多个候选分子结构1402;确定每个候选分子结构的预测时间分辨离子淌度质谱(trim-ms)光谱1404;至少部分地基于分子的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子结构的子集1406;确定子集中的每个候选分子结构的候选分数1408;基于候选分数分配分子的分子结构1410;创建和/或更新分子的数据文件1412;和/或提供分子的分子结构1414。
在实施例中,本公开的方法包括基于分子的路易斯结构确定分子的多个候选分子结构。在实施例中,确定多个候选分子结构可以包括:基于分子的路易斯结构,使用无偏构象搜索确定多个初始候选结构;确定每个初始候选结构的离子化候选结构,其中每个离子化候选结构具有相同的电荷态;以及通过在分子动力学模拟中放松每个离子化候选结构,确定每个离子化候选结构的候选分子结构。
在实施例中,本公开的方法包括确定每个候选分子结构的预测时间分辨离子淌度质谱(trim-ms)光谱。在实施例中,确定每个候选分子结构的预测trim-ms光谱包括确定每个候选分子结构的碰撞截面;和基于碰撞截面,确定每个候选分子结构的预测trim-ms。在实施例中,每个候选分子结构的碰撞截面由投影叠加逼近(psa)或局部碰撞概率逼近(lcpa)确定。在实施例中,候选分子结构的子集包括这样的候选分子结构,即当偏移δω时,其对应的预测trim-ms光谱与测量的trim-ms光谱重叠。
在实施例中,本公开的方法包括至少部分地基于分子的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子结构的子集。
在实施例中,本公开的方法包括确定子集中每个候选分子结构的候选分数。在实施例中,确定子集中每个候选分子结构的候选分数包括对于子集中的每个候选分子结构,确定每个原子-原子对的原子-原子距离概率分布;和对于子集中的每个候选分子结构,通过基于每个原子-原子对的原子-原子距离概率分布的联合概率来确定候选分数。
在实施例中,本公开的方法包括基于候选分数分配分子的分子结构。在实施例中,分配分子的分子结构包括确定分子的分子结构是对应于最高排名候选分数的候选分子结构。
在实施例中,本公开的方法包括创建和/或更新分子的数据文件。在实施例中,日期文件包括分子的标识符;和分子的分子结构。
在实施例中,本公开的方法包括提供分子的分子结构。在实施例中,提供分子的分子结构包括显示分子的分子结构的图像、显示分子的分子结构的坐标、发送分子的分子结构的图像、发送分子的分子结构的坐标或其组合。
本文提供了用于确定分子组合体的分子结构的方法1500,如图15所示。该方法可以包括以下一项或多项:确定分子的多个候选分子组合体1502;确定每个候选分子组合体的预测时间分辨离子淌度质谱(trim-ms)光谱1504;至少部分地基于分子的分子组合体的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子组合体的子集1506;确定子集中每个候选分子组合体的候选分数1508;以及基于候选分数分配分子的分子组合体1510;创建和/或更新分子的新的数据文件1512;和/或提供分子组合体的分子结构1514。
在实施例中,本公开的方法包括确定分子的多个候选分子组合体。在实施例中,确定多个候选分子组合体包括通过大分子对接算法将分子、一个或多个分子组合体中的一个或多个或其组合彼此对接,以形成所述多个候选分子组合体。在实施例中,分子大分子对接算法是zdock或haddock。
在实施例中,本公开的方法包括确定每个候选分子组合体的预测时间分辨离子淌度质谱(trim-ms)光谱。在实施例中,确定每个候选分子组合体的预测trim-ms光谱包括确定每个候选分子组合体的碰撞截面;和基于碰撞截面,确定每个候选分子组合体的预测trim-ms光谱。在实施例中,每个候选分子组合体的碰撞横截面由投影叠加逼近(psa)或局部碰撞概率逼近(lcpa)确定。
在实施例中,本公开的方法包括至少部分地基于分子的分子组合体的测量的trim-ms光谱和每个预测trim-ms光谱之间的相似性度量,确定候选分子组合体的子集。在实施例中,候选分子组合体的子集包括这样的候选分子组合体,即当偏移δω时,其对应的预测trim-ms光谱与测量的trim-ms光谱重叠。
在实施例中,本公开的方法包括确定子集中每个候选分子组合体的候选分数。在实施例中,确定子集中每个候选分子组合体的候选分数包括将分子的分子组合体的测量的trim-ms光谱与每个预测trim-ms光谱交叉相关。在实施例中,确定子集中每个候选分子组合体的候选分数包括确定子集中每个候选分子组合体的概率排名。
在实施例中,本公开的方法包括基于候选分数分配分子的分子组合体。在实施例中,分配分子组合体的分子结构包括确定分子组合体的分子结构是对应于最高排名候选分数的候选分子组合体。
在实施例中,本公开的方法包括创建和/或更新分子的新的数据文件。在实施例中,数据文件包括分子的标识符;和分子组合体的分子结构。
在实施例中,本公开的方法包括提供分子组合体的分子结构。在实施例中,提供分子组合体的分子结构包括显示分子组合体的分子结构的图像、显示分子组合体的分子结构的坐标、发送分子组合体的分子结构的图像、发送分子组合体的分子结构的坐标或其组合。
在实施例中,本公开的方法确定分子或分子组合体的分子结构。在实施例中,分子或分子组合体是有机分子或有机分子组合体。在实施例中,分子或分子组合体是无机分子或无机分子组合体。在实施例中,分子或分子组合体是生物分子或生物分子组合体。在实施例中,分子或分子组合体是蛋白质或蛋白质组合体。蛋白质的基本成分(重复单位)是氨基酸。在实施例中,分子或分子组合体是核酸(例如dna、rna)或核酸组合体。核酸的基本成分(重复单位)是核苷酸(例如脱氧核糖核苷酸、核糖核苷酸)。在实施例中,分子或分子组合体是糖或糖组合体。糖的基本成分(重复单位)是单糖。在实施例中,分子或分子组合体是脂质或脂质组合体。脂质的基本成分(重复单位)是脂肪酸和甘油。
本文提供了药物筛选的方法。该方法可以包括针对根据本公开的实施例确定的分子或分子组合体的分子结构筛选潜在的治疗剂。
图16示出了设备1600或系统的示例的框图,在该设备1600或系统上可以执行本文讨论的任何一种或多种技术(例如,方法)。在其他实施例中,设备1600可以作为独立设备运行,或者可以连接(例如联网)到其他设备。设备1600可以是个人计算机(pc)、台式计算机、移动计算机、膝上型计算机、笔记本计算机、平板计算机、服务器计算机、手持计算机、手持设备或能够执行指令(按顺序或不按顺序)的任何设备,所述指令指定了该设备要进行的动作。此外,虽然仅示出了单个设备,但是术语″设备″还应被认为包括单独或联合执行指令集以执行本文讨论的任何一个或多个方法的任何设备集合,例如云计算或其他计算机集群配置。
如本文所述,示例可以包括逻辑或多个组件、模块或机制,或者可以对其进行操作。模块是能够在运行时执行指定操作的有形实体(例如,硬件)。模块包括硬件。在一个示例中,硬件可以被具体配置为执行特定操作(例如硬连线)。在另一示例中,硬件可以包括可配置的执行单元(例如,晶体管、电路等),和包含指令的计算机可读介质,其中指令配置执行单元以在运行时执行特定操作。配置可以在执行单元或加载机构的指导下进行。因此,当设备运行时,执行单元通信耦合到计算机可读介质。在该示例中,执行单元可以是多于一个模块的构件。例如,在运行时,执行单元可以由第一组指令配置以在一个时间点实现第一模块,由第二组指令重新配置以在第二时间点实现第二模块。
设备1600可以包括处理电路和存储器,用于执行本文描述的操作。在一些实施例中,处理电路可以被配置为执行图14和15中详述的操作。在一些实施例中,设备1600的处理电路可以包括一个或多个处理器。存储器可以存储用于配置处理电路以执行操作的信息,从而执行本文描述的各种操作。存储器可以包括任何类型的存储器,包括非暂时性存储器,用于以机器(例如,计算机)可读的形式存储信息。例如,存储器可以包括计算机可读存储设备、只读存储器(rom)、随机存取存储器(ram)、磁盘存储介质、光存储介质、闪存设备和其他存储设备和介质。
某些实施例可以在硬件、固件和软件中的一种或组合中实现。其他实施例也可以实现为存储在计算机可读存储设备上的指令,所述指令可以由至少一个处理器读取和执行以实现本文描述的操作。计算机可读存储设备可以包括用于以机器(例如,计算机)可读的形式存储信息的任何非暂时性存储机制。例如,计算机可读存储设备可以包括只读存储器(rom)、随机存取存储器(ram)、磁盘存储介质、光存储介质、闪存设备以及其他存储设备和介质。在一些实施例中,设备1600可以包括一个或多个处理器,并且可以配置有存储在计算机可读存储设备存储器上的指令。
设备(例如,计算机系统)1600可以包括硬件处理器1602(例如,中央处理器(cpu)、硬件处理器核或其任意组合)、主存储器1604和静态存储器1606,其中一些或全部可以经由连接件(例如,总线)1608彼此通信。设备1600还可以包括存储设备(即,驱动单元)1610。存储设备1610可以包括机器可读介质1612,其上存储有一组或多组数据结构或指令1614(例如,软件),这些数据结构或指令体现本文描述的任何一种或多种技术或功能或由其使用。在设备1600执行指令1614期间,指令还可以完全或至少部分地驻留在主存储器1604内、静态存储器1606内或硬件处理器1602内。在一个示例中,硬件处理器1602、主存储器1604、静态存储器1606或存储设备1610中的一个或任意组合可以构成机器可读介质。虽然机器可读介质1612被示为单个介质,但是术语″机器可读介质″可以包括被配置为存储一个或多个指令1614的单个介质或多个介质(例如,集中式或分布式数据库,和/或相关联的高速缓存和服务器)。
各种实施例可以完全或部分地以软件和/或固件来实现。该软件和/或固件可以采取包含在非暂时性计算机可读存储介质中或其上的指令的形式。然后,这些指令可以由一个或多个处理器读取和执行,以实现本文描述的操作。指令可以是任何合适的形式,例如但不限于源代码、编译代码、直译码、可执行代码、静态代码、动态代码等。这种计算机可读介质可以包括任何有形的非暂时性介质,用于以一台或多台计算机可读的形式存储信息,例如但不限于只读存储器(rom)、随机存取存储器(ram)、磁盘存储介质、光存储介质、闪存等。
术语″机器可读介质″可以包括能够存储、编码或携带由设备1600执行的指令并且使得设备1600执行本公开的任何一种或多种技术的任何介质,或者能够存储、编码或携带由这些指令使用或与这些指令相关联的数据结构的任何介质。非限制性机器可读介质示例可以包括固态存储器以及光学和磁性介质。在一个示例中,密集机器可读介质(massedmachine-readabiemedia)包括具有多个具有静止质量的粒子的机器可读介质。密集机器可读介质的特定示例可以包括非易失性存储器,例如半导体存储器设备(例如,电可编程只读存储器(eprom)或电可擦除可编程只读存储器(eeprom)和闪存设备;磁盘,例如内部硬盘和可移动磁盘;磁光盘;以及cd-rom和dvd-rom光盘。
以上参考根据各种实施方式的系统、方法、装置和/或计算机程序产品的框图和流程图描述了本公开的某些方面。应当理解,框图和流程图中的一个或多个框以及框图和流程图中的框的组合可以分别由计算机可执行程序指令来实现。同样,根据一些实施方式,框图和流程图中的一些框可能不一定按呈现的顺序执行,或者根本不一定需要执行。
这些计算机可执行程序指令可以被加载到专用计算机或其他特定机器、处理器或其他可编程数据处理装置上以产生特定机器,使得在计算机、处理器或其他可编程数据处理装置上执行的指令创建用于实现流程图方框中指定的一个或多个功能的装置。这些计算机程序指令还可以存储在计算机可读存储介质或存储器中,该计算机可读存储介质或存储器可以指示计算机或其他可编程数据处理装置以特定方式工作,使得存储在计算机可读存储介质中的指令产生包括实现流程图方框中指定的一个或多个功能的指令装置的制品。作为示例,某些实现方式可以提供一种计算机程序产品,其包括计算机可读存储介质,该计算机可读存储介质具有在其中实现的计算机可读程序代码或程序指令,所述计算机可读程序代码适于被执行以实现在流程图框中指定的一个或多个功能。计算机程序指令还可以被加载到计算机或其他可编程数据处理装置上,以使一系列操作元件或步骤在计算机或其他可编程装置上执行,从而产生计算机实现的过程,使得在计算机或其他可编程装置上执行的指令提供用于实现流程图框中指定的功能的元件或步骤。
因此,框图和流程图的框支持用于执行指定功能的装置的组合、用于执行指定功能的元件或步骤的组合以及用于执行指定功能的程序指令装置。还应当理解,框图和流程图的每个框,以及框图和流程图中的框的组合,可以由执行指定功能、元件或步骤的专用、基于硬件的计算机系统,或者专用硬件和计算机指令的组合来实现。
受益于前述描述和相关附图中给出的教导,本文阐述的本公开的许多修改和其他实施方式将是显而易见的。因此,应当理解,本公开不限于所公开的具体实施方式,修改和其他实施方式旨在包括在所附权利要求的范围内。尽管本文使用了特定术语,但是它们仅在一般和描述性的意义上使用,而不是出于限制的目的。
具体示例
ims-ms在阐明分子结构中的适用性
天然蛋白质结构需要在气相中保留足够长的时间,同时充分保持蛋白质的结构完整性,以确保根据ims-ms数据可靠和准确地阐明蛋白质结构。数据表明,蛋白质亚结构之间的协同作用可以在至少几秒钟内防止蛋白质在气相中变性。通过比较由时间分辨ims-ms测量的解折叠速率常数和由蛋白质变体溶液技术测量的解折叠速率常数,可以评估ims-ms数据反映蛋白质天然状态的程度。
图3示出了推进tims-ms用于结构生物学的努力结果。这些努力实现了对测量和时间分辨离子捕获过程的基本理解,测量截面而无需校准的能力,以及研究蛋白质结构和反应性的能力。
ims-ms仪器
ims-ms测量可以在捕获的ims-ms(tims-ms)平台上进行。tims-ms相对于其他ims-ms系统的一个主要优势是其分辨率要高得多。tims-ms提供长达15秒的时间分辨测量。图2示出了根据本公开的实施例的适合使用的双级tims-ms系统。
测定分子截面的方法
psa和lcpa方法准确有效地预测不同缓冲气体中的截面,如图1所示。因此,这些方法对于通过ims-ms重新阐明蛋白质结构是决定性的。用于截面计算的psa和lcpa算法与轨迹法(tjm)一样精确,但速度要快得多。投影逼近是不精确的(约30%误差)。
泛素作为模型分子
本公开定量评估了蛋白质泛素的计算ims-ms数据反映泛素的天然状态(例如,泛素的溶液结构)的程度。这样做是为了测试本公开的系统和方法是否能够根据ims-ms数据准确阐明蛋白质结构,并提供从ims-ms数据中提取生物相关信息的方法。
本公开部分源于这样的理解,即由于不同蛋白质亚结构之间的协同作用,生物相关结构可以在ims-ms实验中保留至少几秒钟。使用泛素作为模型分子,本公开通过使用捕获的ims-ms平台进行的时间分辨测量来研究泛素变体解折叠的动力学和机制,并将这些结果与溶液中的解折叠进行比较(即φ值分析)。
为了研究生理条件下溶液中发生的生物过程,ims-ms测定溶液中蛋白质的天然结构。这些天然蛋白质结构通常包括疏水核心和亲水表面。然而,ims-ms方法测量蛋白质在气相中的截面,其中蛋白质倾向于亲水核心和疏水表面。天然蛋白质结构的某些方面可能被″软″ims-ms仪器动态捕获。对于低电荷态,记录在″软″漂移管上的泛素截面与nmr结构一致。
已经表明,改进的tims-ms仪器重现漂移管数据,低电荷态下的截面与nmr结构一致,如图5所示。本系统和方法可以在与时间分辨tims-ms测量完全相同的位置处捕获泛素离子并对其进行结构表征。本系统和方法可以定量分析时间分辨ims-ms光谱,并且根据本公开的tims-ms仪器可以实现在ims分辨率超过约200的情况下研究气相中的蛋白质解折叠长达15秒。
时间分辨ims-ms光谱,如图6a所示,说明泛素的类天然特征在几秒钟的时间标度上是稳定的。解折叠的速率常数为约0.16s-1(6+)和约o.20s-1(7+),这突出了本公开的设置在保持类天然蛋白质结构方面的能力。这些速率常数定性上与溶液中进行的测量一致。此外,对于电荷态7+,观察到碰撞激活的构象重新折叠成致密结构,随后解折叠成截面与初始构象相差约1%的种类。这一观察非常重要,因为它表明,在ims-ms实验中,通常需要ims≥100才能准确探测生物分析物的动态。该ims分辨率通常在本公开的系统和方法中实现,但一般不在本领域当前已知的其他系统中实现。因此,数据显示,本公开的ims-ms系统能够在″软″条件下以高分辨率在无溶剂的情况下长时间探测蛋白质动态。
在示例性实施例中,本公开的系统和方法预测泛素的每个电荷态的时间分辨ims-ms光谱,以鉴定时间分辨ims-ms实验探测的蛋白质结构的变化,如图6b中对6+所示,而不是比较特定结构时间的各个截面值。
在实施例中,本公开的系统和方法通过基于泛素nmr结构进行显式溶剂分子动力学(md)模拟来预测泛素的ims-ms光谱。md模拟解决了气相中的蛋白质动力学问题。考虑了来自溶液模拟的数千个结构,所有电荷态都被明确处理。随后使用psa和lcpa方法计算解折叠轨迹期间各个步骤的截面,用于与时间分辨ims-ms实验相关联。之所以使用psa和lcpa方法,是因为这两种方法均比较准确且计算效率高。
如图6b所示,泛素电荷态6+的预测时间分辨光谱与实验非常吻合,从而再次确定了预测结构和实验结构之间的一致性。这就实现了在实验中揭示解折叠机制。如图7所示,可以分析气相模拟期间天然接触如何变化。数据表明,当n-端和c-端β-链之间的相互作用消失时,泛素在气相中的变性就开始了。这一观察表明,不同泛素亚结构之间的协同作用阻碍了气相中的变性。
通过比较泛素突变体各自的解折叠速率常数和机制,可以确定气相和溶液相中的协同效应有多密切,如下所示:
(i)蛋白质模型系统使用泛素作为模型系统,因为它已经被包括
(ii)解折叠动力学可以根据时间分辨ims-ms光谱确定,如相对于1-10μm的样品所进行的描述(图6a)。回归分析可以从时间分辨数据中提取解折叠速率常数。每种泛素变体都有不同的速率常数。
(iii)对于(ii)项下研究的每个变体,泛素在气相中的电荷态特异性动力学可以如所述(图6b)进行模拟。通过修饰残基,可以根据wt泛素的nmr和x射线结构来构建初始结构。md模拟可以进行100毫微秒,如上所述。这些模拟的快照可以用于进行如上所述的气相md模拟,但是对于每个电荷态以及opls力场,如初步数据所证明的那样(图6b)。为了获得不同的电荷态,基于半经验am1哈密顿量的静电势计算,氨基酸残基被质子化或去质子化。
(iv)通过psa算法预测ims-ms光谱。碰撞截面可以通过psa方法计算,以预测ims-ms光谱,用于实验的结构分析(图6b)。具体而言,psa计算可以针对从(iii)项下的每个气相解折叠轨迹和质子化状态获得的每个初始和最终结构进行。基于步骤(iii)的气相md模拟,可以对结构进行聚类。每个群集的平均psa截面由该群集的总体加权。数据证明了这种方法的合理性(图6b)。
(v)一旦实验光谱和预测光谱之间达成一致(图6b),解折叠过程的机械解释可以如所述确定(图7)。如所述计算的亲水和疏水表面积的变化可以鉴定解折叠过程中的球状结构变化。天然接触的变化可以鉴定(1)泛素变性的总体程度,(2)泛素亚结构在气相中的相对稳定性,以及(3)不同亚结构之间相互作用的强度(图7)。
步骤(ii)可以确定(i)中表达的泛素变体在气相中的解折叠速率常数。泛素变体显示出不同的解折叠速率常数。这些气相解折叠速率常数可以与溶液中测量的相应解折叠速率常数进行比较。如果溶液和ims-ms中的解折叠是相等的,那么溶液和ims-ms中的解折叠速率可以是正线性相关的。偏离这种预期相关性可以鉴定溶液和ims-ms中结构和解折叠过渡状态之间的差异。不同的突变可以在残基特异性水平上揭示这些差异。步骤(iii)至(v)中的活动确定tims-ms中各种泛素系统的解折叠机制(图6b和图7)。这种分析可以鉴定泛素变体的哪些子结构比其他子结构更容易解折叠,以及各种子结构之间的哪些相互作用保持类天然的时间比其他相互作用长(图7)。总之,本公开的系统和方法可以鉴定溶剂对泛素中的协同效应的影响,从而鉴定ims-ms数据反映泛素天然状态的程度。这一知识可以用于通过ims-ms可靠地阐明蛋白质结构。
本公开的系统和方法可以有效鉴定ims-ms数据反映泛素天然状态的程度。本公开的系统和方法通常也可以应用于其它分子,包括固有无序化蛋白质(idps)、蛋白质低聚物或膜蛋白受体。对固有无序化蛋白质(如乙酰胆碱受体)进行了
在实施例中,本公开提供了用于ims-ms数据分析的自动化软件算法,以便能够重新进行蛋白质结构阐明。在实施例中,本公开的系统和方法不使用基于已知nmr或x射线结构的构象搜索,而是使用没有这种偏差的全面构象搜索,如图8所示。
本系统和方法用于基于泛素nmr结构预测各种泛素电荷态的时间分辨ims-ms光谱。时间分辨实验光谱和预测光谱的比较,如图8所示,显示出高度一致性。数据显示,只要最初的构象搜索样本是天然结构,那么本公开的计算程序就能准确预测蛋白质的时间分辨电荷态特异性ims-ms光谱。此外,时间分辨ims-ms光谱显示,初始实验光谱中的致密峰
重新测定分子结构的示例性方法
根据本发明的示例性实施例,分子结构可以通过以下方式重新鉴定:
(i)测量时间分辨tims-ms光谱,并使用时间分辨光谱来鉴定″天然峰″。
(ii)基于对目标化学结构的认识进行全面、无偏构象搜索。该步骤可以产生大量(>5000)候选结构用于进一步分析。
(iii)电离(例如质子化或去质子化)在(ii)项下产生的候选结构,以获得用作步骤(i)的实验输入的所有电荷态。对于用作输入的每个实验电荷态,该步骤可以为在(ii)项下产生的每个候选结构产生唯一的电荷态。
(iv)通过md模拟放松气相中的带电结构。每20皮秒保存快照一次以供分析。先前的经验表明,这种方法准确地解释了气相中的电荷态特异性分子动力学。可以对从步骤(ii)和(iii)获得的每个结构进行这些计算。
(v)基于步骤(iv)获得的轨迹的psa截面预测时间分辨ims光谱。该步骤预测电荷态特异性ims,用于与步骤(i)中记录的实验进行比较。
(vi)通过交叉相关测量预测和实验离子淌度谱之间的相似性。该方法是合理的,因为数据表明预测光谱中的峰值相对于实验大体上偏移了δω。相似性评分可以计算截面最大偏移(δω),该偏移使实验光谱和预测光谱ei和pi之间的交叉相关xi(δω)=ei*pi最大化。
(vii)如果预测光谱和实验光谱之间的截面最大得分偏移(δω)低于用户定义的阈值,则迭代优化预测ims光谱。为此,步骤(v)和(vi)可以在从预测光谱中去除外围结构之后迭代重复,直到达到期望的阈值。该步骤鉴定来自步骤(i)的初始结构(例如>5000)的子集,该子集最好地解释了实验ims-ms数据。
(viii)基于对步骤(vii)中鉴定的结构的聚类分析,鉴定分子结构。这种结构将反映天然蛋白质结构(图10)。
重新测定分子组合体的示例性方法
在实施例中,本公开提供了一种根据im-ms数据测定蛋白质组合体的结构的软件算法。在实施例中,本公开的系统和方法可以根据动态编程算法进行调整,以计算斐波纳契数(见图12)。第一步是确定蛋白质单体的结构并制成表格(n=1)。然后,策略是迭代地组合较小种类的列表结构{1,...,n-1},以得到更大的组合体(n>1),直到获得所需的组合体(n=m)。例如,列表单体结构(n=1)组合成二聚体(n=2;2=1+1)。然后,三聚体(n=3)由三种单体(3=1+1+1)以及单体和二聚体(3=2+1)构成。四聚体(n=4)由单体、二聚体、三聚体等组合而成。
本公开的系统和方法可以(1)根据im-ms数据鉴定蛋白质单体的结构;(2)由组合较小种类提出(数千)蛋白质组合体结构;(3)计算这些组合体的碰撞截面;(4)根据计算的截面的集合预测im-ms光谱;(5)交叉相关实验和预测质谱,用于(6)组合体结构的概率排名和结果列表。
步骤(1),根据im-ms数据鉴定蛋白质单体的结构,可以通过直方图交叉相关限制分析(搜索)算法的结构说明来进行(图13)。搜索算法可以根据im-ms数据测定蛋白质单体的结构。在步骤(2)中,蛋白质对接方法(例如zdock、haddock)可以用于预测组合体结构。步骤(3)可以通过lcpa方法进行。搜索算法可以采用步骤(4)至(6)。本公开的系统和方法可以阐明具有0至2,000个原子、0至30,000个原子的分子的结构,以及具有多于30,000个原子的结构。
- 还没有人留言评论。精彩留言会获得点赞!