艾维替尼的新合成路线及其马来酸盐二水合物的制作方法
本发明涉及到药用化合物艾维替尼的新合成路线,及其马来酸盐二水合物和相应制剂。
背景技术:
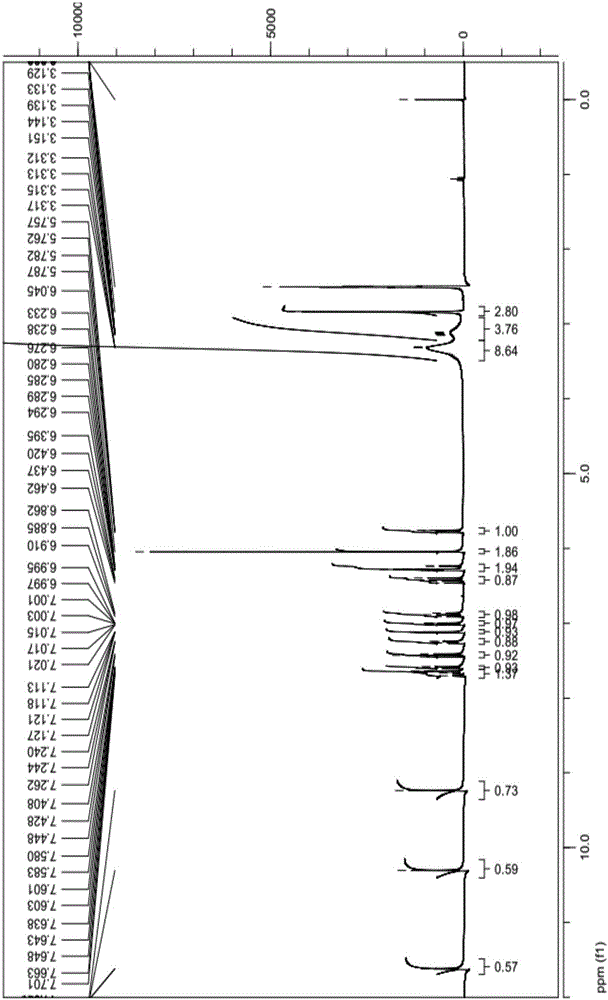
:中国专利cn103748096a号文献公开了艾维替尼化合物及其合成方法。同时,上述专利文献还公开了艾维替尼的马来酸盐和盐酸盐,但是并没有公开马来酸盐的分子结构。根据该专利文献,艾维替尼能够抑制多种激酶,动物试验证明具有抗肿瘤的作用。技术实现要素:本发明的目的是提供上述艾维替尼的新合成工艺。另外,本发明还提供该药用化合物马来酸盐二水合物及其优选晶型,以及以其为活性成分的固体药物制剂。发明人对现有合成路线进行了优化,以2-氯-4-(3-硝基苯氧基)-7氢-(吡咯并[2,3-d]嘧啶)-1-甲基叔丁酸酯为起始原料,以(pd2(dba)3、x-phos作为催化剂,与3-氟-4-(4-甲基哌嗪基)苯胺偶联后,得到2-(3-氟-4-(4-甲基哌嗪基)苯基氨基)-4-(3-硝基苯氧基)-7h-(吡咯并[2,3-d]嘧啶)-1-甲基叔丁酸酯;以甲醇作为溶剂,碱性条件下脱保护基;然后以pd/c为催化剂进行加氢反应,将硝基还原为氨基;最后与丙烯酰氯进行酰化反应,得艾维替尼。本发明还提供艾维替尼的马来酸盐二水合物及其优选晶型。将艾维替尼游离碱化合物和马来酸加至简单醇、乙腈、丙酮、四氢呋喃等有机溶剂或这些有机溶剂的水溶液中,搅拌至澄清,温度控制在25℃~90℃,保温培育5~40min;优选40℃~85℃下保温25~35min。然后进行梯度冷却,得到艾维替尼的马来酸盐二水合物。马来酸与艾维替尼游离碱的当量比为0.9~2:1,优选1~1.2:1。艾维替尼与溶剂的比例为1:10~50(m/v),优选1:20~30(m/v)。其中所述用以成盐的简单醇可以选用甲醇、乙醇、异丙醇等,优选乙醇。药用盐的制备,还可以用任意比例的乙醇/水混合液作为溶剂,优选5%-75%的乙醇/水溶液。在保温过程中会析出淡黄色固体。在搅拌条件下得到的是粉末状结晶;在静置条件下得到的是颗粒状结晶。本发明所述艾维替尼马来酸盐二水合物的结构式如下:本发明马来酸盐二水合物的b晶型和c晶型,具有优异的粉末特性,颗粒大小均匀,这些均匀的小颗粒特别有利于固体制剂加工,特别是制成胶囊剂。马来酸盐二水合物的a晶型、e晶型成颗粒状,在结晶过程中对过滤特别有利。相对而言,a晶型、e晶型的纯度更高。因此,a晶型、e晶型在原料药的提纯和储存过程中会比较有利。通过x射线衍射试验发现,晶型a、b、c、e分别具有如下特征峰:本发明还提供一种以艾维替尼马来酸盐二水合物为活性成分的胶囊剂,所述固体药物制剂还包括如下辅料:硅化微晶纤维素、交联羧甲基纤维素钠、硬脂富马酸钠。硅化微晶纤维素(smcc)是指微晶纤维素与微粉硅胶按照一定重量比例(例如98:2)预先混合在一起制成的商品化药用辅料,例如经过喷雾干燥制成,具有更好的流动性,可以减少辅料的种类,是一种新型药物填料。交联羧甲基纤维素钠、硬脂富马酸钠分别起崩解剂和润滑剂的作用。活性成分和辅料的配比关系如下:附图说明图1是实施例1中所得化合物的氢谱图;图2是实施例2中所得化合物的氢谱图;图3是实施例3中所得化合物的氢谱图;图4是实施例5中所得化合物的氢谱图;图5是实施例3所得化合物的单晶结构图;图6是晶型a、b、c、e的x射线衍射图谱;图6-1至6-4是晶型a、b、c、e的x射线衍射图谱。其中:图6-1颗粒晶型a,图6-2颗粒晶型e,图6-3颗粒晶型b,图6-4颗粒晶型c。具体实施方式第一部分化合物合成1、合成路线a本发明合成路线a中,以2,4-二氯-7h-吡咯-[2,3d]嘧啶为起始原料,硅醚化后,先后引入间硝基苯酚、3-氟-4-甲基哌嗪苯胺,再经还原、丙烯酰氨化,最后脱去引入硅醚保护基团得目标产物。相对于现有合成路线,路线a中第一步用强碱nah,使硅醚化完全反应,第七步用碳酸钾替代三乙胺,由此产率更高,后处理更容易。路线a的具体合成步骤1.12,4-二氯-7-(2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(4)的合成2,4-二氯-7h-吡咯-[2,3d]嘧啶(2,3.70g,19.68mmol)和2-(三甲基硅烷基)乙氧甲基氯(3,3.8ml,21.64mmol)溶于无水二甲基甲酰胺(40ml)形成混合溶液,在0℃搅拌下,加入nah(60%矿物油,945mg,23.61mmol)。反应升温至室温并搅拌2小时,然后用乙酸乙酯(20ml×3)萃取。将有机层合并,用盐水洗涤,经无水硫酸钠干燥并过滤。浓缩滤液,粗品经色谱法纯化,得到5.66g2,4-二氯-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶为无色液体,产率为90.4%。1hnmr(cdcl3,400mhz):7.37(1h,d,4hz),6.66(1h,d,4hz),5.60(2h,s),3.55(2h,t,8hz),0.94(2h,t,8hz),0.02(9h,s);13cnmr(cdcl3,100mhz):154.2,130.9,117.9,102.5,74.8,68.5,19.1,0.05.1.22-氯-4-(3-硝基苯氧基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(6)的合成2,4-二氯-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(4,5.4g,16.95mmol)和3-硝基苯酚(5,2.59g,18.68mmol)溶于50mldmf形成混合物,加入k2co3(4.68g,37.36mmol),反应混合物升温至室温,并搅拌3小时,加入水稀释,用乙酸乙酯(40ml×3)萃取。将有机层合并,用盐水洗涤,经无水硫酸钠干燥并过滤,浓缩滤液得粗品,经色谱法纯化,得到6.71g2-氯-4-(3-硝基苯氧基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(6)为白色固体,产率94.3%.1hnmr(cdcl3,400mhz):8.15(2h,s),7.64(2h,d,8hz),7.30(1h,d,4hz),6.65(1h,d,4hz),5.61(2h,s),3.58(2h,t,8hz),0.94(2h,t,8hz),0.02(9h,s);1hnmr(cdcl3,100mhz):155.2,131.6,129.6,129.0,122.1,118.7,101.3,74.8,68.5,19.1,0.0.1.3n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-4-(3-硝基苯氧基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-2-胺(8)的合成将2-氯-4-(3-硝基苯氧基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶(6,4.5g,10.68mmol)、4-(4-甲基哌嗪基苯胺(7,2.1g,10.68mmol)、三(二亚苄基丙酮)二钯(pd2(dba)3,1.08g,1.068mmol),二环已基(2’4’6’-三异丙基联苯-2-膦)(x-phos,3g,6.42mmol)和碳酸钾(6g,5.27mmol)溶解在叔丁醇(200ml),在氮气下、80℃搅拌过夜。冷却至室温后,将反应混合物通过硅藻土垫过滤,乙醇洗涤。得5.64g淡黄色泡沫固体n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-4-(3-硝基苯氧基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-2-胺(8)。1hnmr(cdcl3,400mhz):8.20(2h,m),7.63(2h,t,4hz),7.51(1h,d,8hz),7.03(1h,d,4hz),6.90(1h,t,8hz),6.81(1h,t,8hz),6.52(1h,d,4hz),5.52(2h,s),3.62(2h,t,8hz),3.04(4h,t,8hz),2.61(4h,t,8hz),2.45(3h,s),0.95(2h,t,8hz),0.02(9h,s);13hnmr(cdcl3,100mhz):163.2,156.7,156.3,155.7,154.8,150.3,137.1,136.9,135.6,135.5,131.5,130.1,125.9,121.7,120.5,120.4,119.3,115.3,108.7,108.4,101.3,100.8,74.4,67.8,56.6,52.1,51.7,47.5,19.1,0.0.1.44-(3-氨基苯氧基)-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-2-胺(9)的合成将n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-4-(3-硝基苯氧基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-2-胺(8)(5.9g,10.0mmol)溶于60ml乙醇与10ml水的混合液中,然后分别加入铁粉(2.8g,50.0mmol)和氯化铵(5.34g,0.1mol),在氮气下回流搅拌2小时。冷却至室温后,将反应混合物通过硅藻土垫过滤,乙醇洗涤,经柱层析(甲醇:二氯甲烷=1:16(v/v),得5.5g4-(3-氨基苯氧基)-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-2-胺(9),产率98.2%。还原也可以用pd/c氢化,几乎定量转化。1hnmr(cdcl3,400mhz):7.67(2h,d,12hz),7.21(1h,t,8hz),7.05(1h,d,8hz),6.95(1h,s),6.92(1h,s),6.88(1h,t,8hz),6.81(1h,t,8hz),6.65(3h,m),6.32(1h,d,4hz),5.52(2h,s),3.60(2h,t,8hz),3.08(4h,t,8hz),2.64(4h,t,8hz),2.39(3h,s),0.92(2h,t,8hz),0.02(9h,s).13hnmr(cdcl3,400mhz):172.6,164.3,158.4,156.6.156.0,155.6,149.3,137.8,137.5,135.2,131.6,125.2,120.7,115.1,113.8,110.2,108.5,101.8,100.9,74.4,67.8,61.8,56.6,52.2,47.5,41.7,22.5,19.2,15.7,0.0.1.5n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(10)合成将(3-氨基苯氧基)-n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-2-胺(9)(5.6g,10.0mmol)溶于100ml无水二氯甲烷中,随后加入二异丙基乙胺(3.3ml,20mmol)冷却至0℃,10分钟内滴加丙烯酰氯(1.2ml,15mmol),撤去冷源,室温搅拌2小时,加乙酸乙酯150ml稀释,加入100ml水终止反应,水层用100ml乙酸乙酯萃取,合并有机层,浓缩得粗品。经柱层析(甲醇:二氯甲烷=1:15(v/v)),得5.24g乳白色泡沫固体n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(10),产率85%。1hnmr(cdcl3,400mhz):7.75-7.57(3h,m),7.00(2h,s),6.82(2h,s),6.82(1h,t,8hz),6.40(2h,d,4hz),6.29(1h,m),5.73(1h,d,12hz),5.51(2h,s),3.60(2h,t,8hz),3.05(4h,t,8hz),2.65(4h,t,8hz),2.38(3h,s),0.96(2h,t,8hz),0.0(9h,s).1.6n-(3-(2-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-(羟基甲基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(11)合成在0℃下,将n-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-((2-(三甲基硅烷基)乙氧基)甲基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(10)(3.09g,5.0mmol)溶于50ml无水二氯甲烷和17mlcf3cooh混合液中,tlc监控反应进程,3小时反应完成,加入碳酸氢钠饱和溶液中和三氟乙酸(防止大量气饱冒出),再用水洗至中性,再用100ml乙酸乙酯萃取,合并有机层,浓缩得粗品。经柱层析(甲醇:二氯甲烷=1:15(v/v),得2.34g乳白色泡沫固体n-(3-(2-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-(羟基甲基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(11),产率90.5%。1hnmr(dmso,400mhz):10.33(1h,s),9.31(1h,s),8.31(1h,s),7.69(1h,s),7.61-7.42(3h,m),7.30(1h,8hz),7.22(1h,d,8hz),7.01(1h,d,8hz),6.79(1h,t,12hz),6.44-6.37(2h,m),6.28-6.23(1h,d,12hz),5.78(1h,d,8hz),5.49(2h,s),2.88(4h,s),2.44(4h,s),2.21(3h,s).13hnmr(dmso,100mhz):163.7,162.4,155.3,154.4,153.9,153.5,140.8,136.9,133.5,132.2,130.4,127.7,125.2,119.4,117.3,116.6,114.4,113.3,106.6,99.2,67.00,55.3,50.9,46.3.1.7n-(3-(2-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(1)合成在室温下,将n-(3-(2-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7-(羟基甲基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(11)(5.17g,10.0mmol)溶于100ml无水乙醇中,加入无水碳酸钾(4.14g,30mmol),tlc监控反应进程,4小时左右后反应结束,过滤,低温浓缩去除乙醇得粗品。经柱层析(甲醇:二氯甲烷=1:15(v/v),得4.23g淡黄色泡沫固体n-(3-(2-(3-氟-4-(4-甲基哌嗪-1-基)苯基)-7h-吡咯并[2,3-d]嘧啶-4-基氧)苯基)丙烯酰胺(1),产率86.7%。1hnmr(dmso,400mhz):11.61(1h,s),10.31((1h,s),9.15(1h,s),7.67(1h,s),7.59(2h,d,8hz),7.44-7.40(1h,m),7.24(1h,m),7.10(1h,t,4hz),7.01(1h,d,8hz),6.82(1h,t,8hz),6.46-6.39(1h,m),6.29(2h,t,4hz),5.78(1h,d,12hz),2.88(4h,t,8hz),2.45(4h,t,8hz),2.22(3h,s).13hnmr(dmso,100mhz):163.7,162.3,155.4,155.6,155.3,153.9,153.6,140.8,133.4,132.2,130.3,127.7,122.4,119.4,117.3,116.5,114.4,113.2,106.6,99.0,55.3,50.9,46.3.2、合成路线b发明人还发明了另外一条合成路线,即路线b,其合成步骤具体描述如下。2.12-(3-氟-4-(4-甲基哌嗪基)苯基氨基)-4-(3-硝基苯氧基)-7h-(吡咯并[2,3-d]嘧啶)-1-甲基叔丁酸酯(2b)的合成在常温氮气保护下,将4.05g2-氯-4-(3-硝基苯氧基)-7氢-(吡咯并[2,3-d]嘧啶)-1-甲基叔丁酸酯(3b,0.01mol)溶于200ml叔丁醇中,磁力搅拌溶解后,依次加入三(二亚苄基丙酮)二钯(pd2(dba)3,0.186g)、2-二环己基磷-2,4,6-三异丙基联苯(x-phos,0.19g)、3-氟-4-(4-甲基哌嗪基)苯胺(4b,2.30g,0.011mol)、无水碳酸钾(6.9g,0.05mol),继续搅拌10分钟后,升至80℃,反应持续5小时。tlc检测反应进程,反应结束后,反应液降至常温,用硅藻土过滤,乙酸乙酯洗涤,滤液减压浓缩得7.8克粗产品,经硅胶柱(乙酸乙酯:石油醚=1:1(v/v))分离纯化得5.2g克浅黄色固体:2-(3-氟-4-(4-甲基哌嗪基)苯基氨基)-4-(3-硝基苯氧基)-7h-(吡咯并[2,3-d]嘧啶)-1-甲基叔丁酸酯(2b),产率90.2%。1hnmr(400mhz,cd3od),δ=8.18-8.13(m,2h),7.67-7.64(m,2h),7.42(d,j=15.2hz,1h),7.13(t,j=3.6hz,1h),7.01(d,j=8hz,1h),6.77(t,j=9.2hz,1h),6.49(t,j=3.6hz,1h),6.11(s,2h),2.98(s,4h),2.58(s,4h),2.32(s,3h),1.15(s,9h).2.2n-(3-氟-4-(4-甲基哌嗪基)苯基)-4-(3-硝基苯氧基)-7h-(吡咯并[2,3d]嘧啶)-2-胺(5b)的合成在低温(5~10℃)氮气保护下,将5.78g2-(3-氟-4-(4-甲基哌嗪基)苯基氨基)-4-(3-硝基苯氧基)-7氢-(吡咯并[2,3-d]嘧啶)-1-甲基叔丁酸酯(2b,0.01mol)溶于200ml甲醇溶液中,磁力搅拌溶解后,缓慢滴加2mnaoh溶液(200ml),继续搅拌2小时后,tlc监测进程。反应结束后,反应液先后加入乙酸乙酯与冰水,取有机层,水层用乙酸乙酯再萃取二次,合并有机层,减压浓缩得粗产品,经硅胶柱(乙酸乙酯:石油醚=3:2(v/v))分离纯化得淡黄色固体4.54g克,产率98.1%。1hnmr(400mhz,cd3od),δ=9.76(s,1h),8.21-8.10(m,2h),7.67-7.54(m,2h),7.40(dd,j=15.2,3.0hz,1h),6.96(s,1h),6.89-6.83(m,2h),6.76(d,j=9.0hz,1h),6.46(dd,j=3.6,1.8hz,1h),3.05(s,4h),2.62(s,4h),2.36(s,3h).2.34-(3-氨基苯氧基)-n-(3-氟-4-(4-甲基哌嗪基)苯基)-7h-(吡咯并[2,3-d]嘧啶)-2-胺(6b)的合成在常温常压保护下,将4.63gn-(3-氟-4-(4-甲基哌嗪基)苯基)-4-(3-硝基苯氧基)-7氢-(吡咯并[2,3-d]嘧啶)-2-胺(5b,0.01mol)溶于100ml四氢呋喃溶液中,磁力搅拌溶解后,加入200mg10%钯/碳(pd/c)催化,反应瓶置换成氢气环境,继续搅拌0.5小时后,tlc监测进程,反应结束后,用硅藻土过滤,四氢呋喃洗涤二次,合并滤液减压浓缩,几乎定量转化,无需进行任何后处理,直接用于下一步反应。2.4n-3-(2-(3-氟-4-(4-甲基哌嗪基-1-基)苯基)-7h-(吡咯并[2,3-d]嘧啶)-4-基氧)苯基)丙烯酰胺(1)的合成在氮气保护下,将4.33g4-(3-氨基苯氧基)-n-(3-氟-4-(4-甲基哌嗪基)苯基)-7氢-(吡咯并[2,3d]嘧啶)-2-胺(6b,0.01mol)溶于150ml无水四氢呋喃溶液中,磁力搅拌溶解后,加入5.52g无水碳酸钾(0.04mol),在冰浴条件下,5分钟内滴加1.06ml丙烯酰氯(0.013mol),继续搅拌半小时后升至室温,tlc监测进程,大约反应2小时。如有需要,可以用适量氢氧化钠淬灭反应。反应液过滤除去过量无水碳酸钾,滤液中加入乙酸乙酯和水,有机层用饱和食盐水干燥,减压浓缩得油状粗品5.1克,后者经硅胶柱层析得3.70克浅黄色固体(1),产率76%。1hnmr(400mhz,dmso):11.31(s,1h),10.31(s,1h),9.15(s,1h),7.67(s,1h),7.59(d,j=8.0hz,2h),7.44-7.40(m,2h),7.10(t,j=4.0hz,1h),7.01(d,j=8.0hz,1h),6.82(t,j=8.0hz,1h),6.46-6.39(m,1h),6.29(d,j=4.0hz,1h),5.78(d,j=12.0hz,1h),2.88(s,4h),2.45(s,4h),2.22(s,3h).13cnmr(100mhz,dmso):163.7,162.3,155.6,155.4,155.3,153.9,153.6,140.8,133.4,132.2,130.3,127.7,122.4,119.4,117.3,116.5,114.4,113.2,106.7,98.9,55.3,50.9,46.3.与文献报道的合成路线比较,合成路线b的优势有三:1)反应路线短,总产率高达67.2%,而文献报道产率仅为40%,高出27%。文献报道的路线为五步反应。本发明中钯碳催化反应几乎定量,无需后处理,直接用于下步反应,故上述2.3和2.4可视为一步反应。因此,本发明路线实际上缩为三步,较文献报道的反应路线有所缩短。2)脱保护基更加容易,后处理简单。保护基叔丁酸酯甲基比2-(三甲基甲硅烷基)乙氧甲基更容易脱去,只需用氢氧化钠常温下短时间一步脱掉,而2-(三甲基甲硅烷基)乙氧甲基需要大量无水三氟乙酸和氨水,分二步发生,增加成本,严重限制反应的工艺化。3)现有方法中尽管使用了廉价的铁粉与盐酸,但工业化生产中铁粉处理困难,大量的强酸容易污染环境。因此,本发明合成路线在规模化生产中具有重要的应用价值。第二部分成盐以及晶型1、盐的制备将艾维替尼游离碱化合物和马来酸在乙醇或乙腈的水溶液中进行反应,在35~90℃范围内进行保温培育,然后进行梯度冷却,得到艾维替尼的马来酸盐二水合物。在搅拌条件下进行保温培育的话,得到的是粉末状结晶b和c;在静置条件下得到的是颗粒状结晶a和e。实施例1:马来酸盐(粉末晶型c)化合物艾维替尼520mg(1.07mmol)置于圆底烧瓶,加入7.5ml5%乙醇,40℃下搅拌成糊状。加入固体马来酸148.8mg(1.2eq,1.28mmol),40℃下持续搅拌,糊状物逐步澄清。2分钟后出现淡黄色粉末状固体,且逐步增加。搅拌状态下自然冷却至室温,转至4℃冰箱中静置12h。析出粉末状固体,抽滤,用0℃的5%乙醇洗涤。50℃真空干燥,得淡黄色粉末固体506mg(0.791mmol,74%)。m.p.:180-183℃。1h-nmr鉴定:1h-nmr(400mhz,dmso-d6):δ11.62(1h,brs,nh),10.31(1h,brs,nh),9.24(1h,brs,nh),7.64-7.70(2h,m,ph-h),7.59(1h,dd,j=8.0,1.2hz,ph-h),7.43(1h,t,j=8.0hz,ar-h),7.25(1h,dd=8.8,1.6hz,ph-h),7.12(1h,dd=3.6,2.4hz,ph-h),7.01(1h,ddd=8.0,2.4,0.8hz,ph-h),6.89(1h,t,j=9.6hz,ar-h),6.43(1h,dd=16.8,10.0hz,=ch),6.23-6.29(2h,m,=ch),6.05(2h,brs,ma-h×2),5.77(1h,dd,j=10.0,2.0hz,=ch),3.31(8h,brs,ch2×2,h2o×2),3.13(4h,brs,ch2×2).实施例2:马来酸盐(颗粒晶型e)化合物艾维替尼173.5mg(0.356mmol)置于圆底烧瓶,加入2ml90%乙醇,80℃下搅拌成糊状。加入固体马来酸49.6mg(1.2eq,0.427mmol),搅拌至澄清,持续搅拌30分钟。停止搅拌,自然冷却至室温,再转至4℃冰箱中静置12h。析出淡黄色颗粒状固体,抽滤,用0℃的90%乙醇洗涤。冷冻干燥过夜,得淡黄色颗粒状固体122mg(0.191mmol,53%)。m.p.180-183℃。由于晶型e颗粒较大,抽滤过程非常方便。1h-nmr鉴定:1h-nmr(400mhz,dmso-d6):δ11.65(1h,brs,nh),10.34(1h,brs,nh),9.26(1h,brs,nh),7.67-7.72(2h,m,ph-h),7.61(1h,d,j=8.4hz,ph-h),7.44(1h,t,j=8.0hz,ar-h),7.27(1h,d,j=8.8hz,ph-h),7.13(1h,dd=3.6,2.4hz,ph-h),7.03(1h,dd=8.4,2.4hz,ph-h),6.90(1h,t,j=9.4hz,ar-h),6.45(1h,dd=16.8,10.0hz,=ch),6.25-6.31(2h,m,=ch),6.08(2h,brs,ma-h×2),5.78(1h,dd,j=10.4,1.8hz,=ch),3.14-3.33(12h,brs,ch2×4,h2o×2),2.87(3h,brs,n-ch3).实施例3:马来酸盐(颗粒晶型a)称量化合物艾维替尼220mg(0.45mmol),加至5ml乙腈中,65℃下搅拌至澄清。马来酸63mg(1.2eq,0.54mmol)溶于0.6ml水中,逐滴加至艾维替尼的乙腈溶液中,成澄清的混合液,65℃下搅拌0.5h。停止搅拌,冷却至室温,转至4℃冰箱中静置12h。析出黄色颗粒状固体,抽滤,用5ml0℃的5%乙腈润洗。50℃真空干燥12h,得黄色颗粒固体180mg(0.28mmol,62.5%)。m.p.:181-183℃。由于晶型a颗粒较大,抽滤过程非常方便。1h-nmr鉴定:1h-nmr(400mhz,dmso-d6):δ11.64(1h,brs,nh),10.33(1h,brs,nh),9.25(1h,brs,nh),7.66-7.71(2h,m,ph-h),7.60(1h,dd,j=8.0,1.2hz,ph-h),7.44(1h,t,j=8.0hz,ar-h),7.27(1h,dd=8.4,1.2hz,ph-h),7.13(1h,dd=3.6,2.4hz,ph-h),7.02((1h,ddd,j=8.0,2.4,0.8hz,ph-h),6.90(1h,t,j=9.4hz,ar-h),6.44(1h,dd=16.8,10.0hz,=ch),6.24-6.31(2h,m,=ch),6.07(2h,brs,ma-h×2),5.78(1h,dd,j=10.0,2.0hz,=ch),3.32(4h,brs,ch2×2),3.13(4h,brs,ch2×2),2.85(3h,brs,n-ch3)。通过比较实施例1和实施例3的1h-nmr,我们发现二者化合物结构对应的氢谱是一致的,不同之处是残留的不同溶剂峰:实施例1中残留的是乙醇,在1.06和3.44处的小峰,比例很少,未进行积分。实施例3中的样品残留的是乙腈,积分后显示为0.24,但这不影响化合物的一致性。实施例4:马来酸盐(颗粒晶型a)称量化合物220mg(0.45mmol),加至4.5ml乙腈中,40℃下搅拌至澄清。马来酸63mg(1.2eq,0.54mmol)溶于0.5ml水中,逐滴加至艾维替尼的乙腈溶液中,40℃下搅拌5分钟,出现粉末状固体。升温至70℃,搅拌20分钟,固体溶解后停止搅拌,自然冷却至室温,出现少量颗粒状晶体,转至0℃冰箱中静置12h。析出较多颗粒状固体,抽滤,用5ml0℃的5%乙腈润洗。50℃真空干燥12h,得黄色固体203mg(0.317mmol,70.2%)。m.p.:181-183℃。各项参数和实施例3基本一致。一般纯净的化合物的熔点的起点和终点之间是3℃。颗粒晶型a的熔程是2℃左右,融化起点和终点清晰可见,说明纯度更高,体现在dsc中的峰更加尖锐。其他样品虽然也能基本满足熔点的起点和终点之间是3℃这个条件,但在显微镜法检测时,发现起点不是很清晰,一些颗粒极细小的粉末会先消失,不像晶体颗粒那样整齐地、容易发现地发生融化,需要重复一次才能准确判断其起点温度。另外,相对而言,发明人还发现,乙腈/水结晶体系重复性更好。实施例5:马来酸盐(粉末晶型b)称量化合物220mg(0.45mmol),加至9ml乙腈中,40℃下搅拌至澄清。马来酸63mg(1.2eq,0.54mmol)溶于1ml水中,逐滴加至艾维替尼的乙腈溶液中,40℃下搅拌5分钟,出现粉末状固体。持续搅拌,自然冷却至室温,出现粉末状固体,转至0℃冰箱中静置12h。析出更多粉末固体,抽滤,用5ml0℃的5%乙腈润洗。50℃干燥12h,得黄色固体173mg(0.270mmol,59.8%)。m.p.:181-183℃。1h-nmr鉴定:1h-nmr(400mhz,dmso-d6):δ11.64(1h,brs,nh),10.33(1h,brs,nh),9.25(1h,brs,nh),7.66-7.71(2h,m,ph-h),7.60(1h,dd,j=8.0,1.2hz,ph-h),7.44(1h,t,j=8.2hz,ar-h),7.26(1h,dd=8.8,1.6hz,ph-h),7.13(1h,dd=3.6,2.4hz,ph-h),7.02(1h,ddd,j=8.0,2.4,0.8hz,ph-h),6.90(1h,t,j=9.4hz,ar-h),6.44(1h,dd=16.8,10.0hz,=ch),6.24-6.30(2h,m,=ch),6.07(2h,brs,ma-h×2),5.78(1h,dd,j=10.0,2.0hz,=ch),3.33(8h,brs,ch2×2,h2o×2),3.14(4h,brs,ch2×2),2.85(3h,brs,n-ch3)。实施例6:盐酸盐精密称量化合物311.2mg(0.638mmol)置于圆底烧瓶,加入4ml5%乙醇,40℃搅拌成糊状。将0.117ml37%的盐酸加至1ml5%乙醇中,混匀后滴加至反应瓶中。搅拌至澄清,保温1h。自然冷却至室温,少量粉末固体析出,转至0℃冰箱中静置12h。析出更多固体,抽滤,用0℃的5%乙醇洗涤。冷冻干燥过夜,得灰白色粉末固体201mg(0.337mmol,53%)。2、理化性质2.1加速试验2l的密闭干燥器底部放置250ml饱和na2hpo4溶液(20℃、98%湿度);用25×25mm的称量瓶,称量240mg左右干燥至恒重的盐,置于隔板上。分别在12h、24h、36h和48h时称重,吸湿率变化如表1:表1.在20℃、98%湿度的吸湿率由此可见,游离碱化合物的吸湿性较强,盐酸盐更强,都不太适合作为原料药。相对而言,马来酸盐(晶型a、b、c、e)的吸湿率有相当程度的降低,更加适宜作为原料药。2.2粒径结晶过程中的一个重要方面是晶体的粒径及其分布,需要满足药物生物利用度的要求。对于溶解度差的化合物例如艾维替尼而言,较大的表面积从而获得较大溶出速率,通常为改善生物利用度的关键因素,因此较小的晶体粒径是必要的。但是,通过结晶得到满意的均匀粒径并不容易。而对化合物进行研磨是一种昂贵且高耗能的操作。此外,在通常情况下,经研磨的颗粒的粒径分布并不均匀、粉末流动性差、堆密度低,这导致粉末处理、制剂掺合难以进行,且药物产品的含量均匀性较差。发明人发现,在马来酸盐的结晶过程中,是否搅拌对晶体的粒径有很大的影响。具体而言,在保温培育过程中,如果同时进行持续搅拌的情况下,得到的是粉末状的或颗粒很小的晶体;如果保温培育过程中中反应体系处于静置状态下,则得到较大颗粒状的结晶颗粒。对本发明得到的各种晶型的粒径分布进行了初步测量,结果如下:表2.各种晶型的粒径分布对晶型b和晶型c进行、过筛粉碎,发现很容易将其中的大颗粒粉碎到100μm以下的粒径,从而进一步提高了其粒径的细度和均匀度。3、结晶水分子的测定实施例1-5制备的所有马来酸盐样品经1h-nmr鉴定,确认含有1分子的马来酸,同时含有2分子的结晶水。经过热重tga(自由碱、晶型a和晶型c)和热差dsc(晶型c和晶型d)分析,确认晶型a和晶型c均含有结晶水,结晶水与化合物比例是2:1,不同的晶型失水的温度有所不同。颗粒晶型a在105℃时失重6.31%,90-110℃区间内的曲线斜率较大,失重较快,推测是结晶水,数量为2。粉末晶型c在84.5℃时失重6.49%,80-90℃区间内的曲线斜率较大,失重较快,推测是结晶水,数量为2。181.3℃对应的失重是盐融化过程中的失重,dsc给出的熔点181.3℃与显微镜法测得的180-183℃是一致的。为了更准确地确定结晶水个数,按照实施例3的方法,培养得到单晶,采用ccd-单晶x-射线衍射仪(rigakusaturn724)测试单晶结构。经过解析,该化合物中含有1分子的艾维替尼,1分子的马来酸,2分子的结晶水,结构图如图5所示。4、xrd测定对实施例1-5得到的4个样品a-e进行x射线衍射测试,所得xrd图谱如图6所示,各样品对应的xrpd衍射峰数据如下。表3.样品a的xrpd衍射峰数据表4.样品b的xrpd衍射峰数据表5.样品c的xrpd衍射峰数据表6.样品e的xrpd衍射峰数据将a-e四种粉末的xrd图叠加处理,可以看出,同样是90%乙腈/水结晶体系,静止条件下得到的颗粒状固体(a)和搅拌条件下得到的粉末(b)的图谱明显存在不同,主要区别之处在于θ=25.48处,样品a的吸收峰值为19068,而粉末b在θ=25.40处的吸收峰值为3031;样品a在θ=38.8和46.9处分别有值为2588和2505的吸收峰,而样品b没有这两个吸收峰。颗粒状晶体a和e的图谱比较接近,粉末b与粉末c的图谱较为接近。第三部分胶囊剂如前所述,本发明马来酸盐二水合物的b晶型和c晶型,具有优异的粉末特性,颗粒大小均匀,有利于制成胶囊剂。发明人通过对晶型b和晶型c的粒径及其分布进行深入研究,制定了艾维替尼的胶囊剂处方,具体如下。硅化微晶纤维素(smcc)可以通过商业渠道获得,例如德国jrs公司生产的优化型微晶纤维素prosolv50和prosolv90,主要参数如下:根据处方称取活性成分和辅料(润滑剂硬脂富马酸钠除外),分别过筛粉碎,充分混合,然后再加入润滑剂硬脂富马酸钠混匀,灌装胶囊。胶囊剂制备例1处方胶囊内容物总重200mg,下同。每粒胶囊含51mg活性成分。胶囊剂制备例2处方:以晶型c为活性成分(代替晶型b),各种辅料成分和含量和制备例1相同。胶囊剂制备例3处方胶囊剂制备例4处方:以晶型c为活性成分(代替晶型b),各种辅料成分和含量和制备例3相同。胶囊剂制备例5处方胶囊剂制备例6处方:以晶型c为活性成分(代替晶型b),各种辅料成分和含量和制备例5相同。采用物料流动性测试仪考察胶囊内容物的流动性。以上制备例1-6的休止角都在31度以内,完全可以满足胶囊剂生产过程中的流动性需求。按照中国药典转篮法测定释放度,胶囊制备例1-6的结果详见下表:时间(h)参考标准测定值范围1(h)---3~6%2(h)12-39%24~32%3(h)44-70%49~58%5(h)>70%71~82%当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1