一种阳离子脂质分子及其在核酸递送中的应用的制作方法
本发明涉及一种阳离子脂质分子及其在核酸递送中的应用,属于核酸传递系统领域。
背景技术:
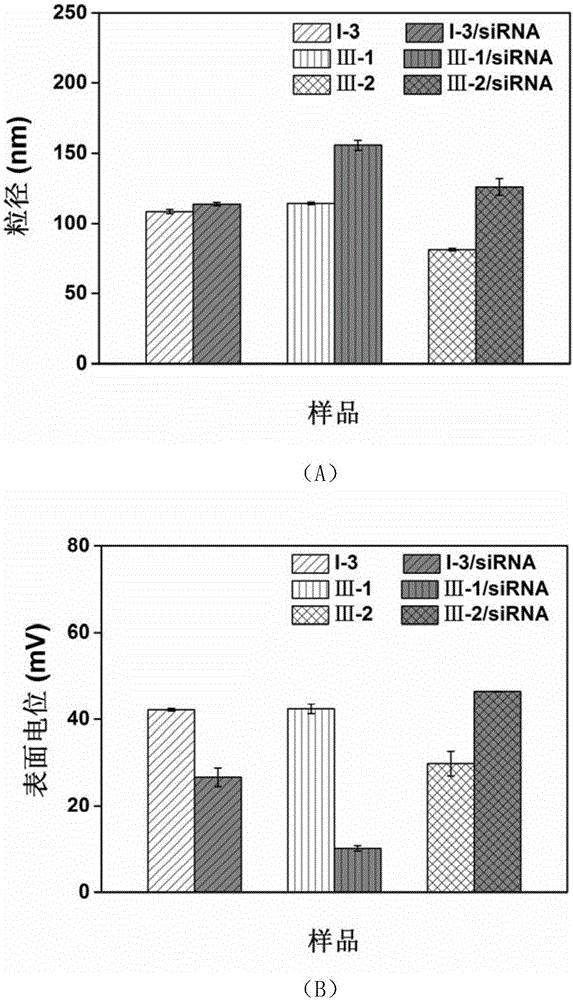
:基因治疗是将外源正常基因导入靶细胞,用以纠正或补偿基因缺失或异常所引起的疾病,从而达到治疗的目的。基因沉默技术是其中的典型代表。为了实现基因沉默,介导分子如sirna必须要进入到靶细胞中并发挥作用。然而,裸sirna易降解,在血浆中的半衰期不到一小时;且由于尺寸非常小,在循环中会迅速被肾脏排出体外;另外sirna不能穿过细胞膜。因此,为了实现sirna的基因沉默功能,需要寻找有效的递送系统保护sirna,避免降解,同时增加细胞的摄入等。阳离子脂质体是一类极具潜力的核酸分子递送载体材料,但常规脂质体的合成通常需要单独优化,且步骤繁琐,合成成本相对困难和昂贵,常常导致对使用者而言价格过高。另外现有的商业化阳离子脂质体仍面临安全性的问题和高效性的挑战。因此,开发具有高效率、低毒性、制备简便可行、性质稳定的新型阳离子脂质体成为了亟待解决的问题。技术实现要素:本发明的目的是提供一种新型阳离子脂质分子及其在核酸递送等方面的应用,所提供的阳离子脂质分子可将活性物质(如sirna)高效递送到细胞(如示例的肺癌细胞)、组织、器官中,实现活性物质的高效调控。本发明所提供的阳离子脂质分子,其结构式如式ⅰ所示:式ⅰ中,a表示c14-28酰基或c14-28烷氧基;b表示或不存在,其中j为1~5之间的整数,m为1~4之间的整数,n为2~4之间的整数;c表示其中k为2~4之间的整数;d表示其中p为1~4之间的整数;e表示其中r1和r2独立地表示c1-4烷基或r1和r2稠合形成任意取代的c5-7杂环基,x为氯、溴或碘原子。式中表示不同位置取代基的吡啶季铵盐,x为氯、溴或碘原子,其包括具体地,式ⅰ所示化合物可为表1中的下述结构:表1式i-ⅶ的结构式其中,a为任选地被插入0个、1个或多个双键的c14-28酰基或c25-27烷氧基,c14-28酰基具体包括但不限于:肉豆蔻油酰基(c14:1,顺-9)、棕榈油酰基(c16:1,顺-9)、油酰基(c18:1,顺-9)、花生四烯酰基(c20:4,顺-5,8,11,14)、芥酰基(c22:1,顺-13)、神经酰基(c24:1,顺-15)、肉豆蔻酰基、棕榈酰基、硬脂酰基、二十碳酰基、二十二碳酰基、二十四碳酰基、二十六碳酰基或二十八碳酰基。本发明提供的阳离子脂质分子具体如表2中所示:表2各阳离子脂质分子的结构式本发明提供的阳离子脂质分子均能通过现有技术中的常规方法进行制备。本发明进一步提供了一种阳离子脂质体,由所述阳离子脂质分子或由所述脂质分子和辅脂制成,所述辅脂为中性脂质分子和/或peg脂质分子;优选的,所述中性脂质分子可为dope;所述peg脂质分子可为dspe-peg。所述阳离子脂质分子与所述中性脂质分子可以等摩尔比混合;所述阳离子脂质分子与所述peg脂质分子的摩尔比可为9:1~2。所述阳离子脂质体为表面带有净正电荷的纳米颗粒。所述阳离子脂质体中,所述阳离子脂质分子的摩尔含量可为30%~70%,优选45%~50%。所述阳离子脂质体至少具有(1)和(2)特征之一:(1)粒径为:80~200nm、89~130nm、100~200nm、80~120nm、80~100nm或80~89nm;(2)表面电位为:5~70mv、30~70mv、30~45mv、5~45mv或5~30mv。本发明所述阳离子脂质体均一且稳定、可作为载体系统使用,如基因载体、转染试剂。本发明阳离子脂质体的制备方法包括但不限制于超声法、反相蒸发法、注入法、冷冻干燥法和高压均质、旋转蒸发法、乳化法等方法。优选的,乳化法在获得透明乳液后还经过了过滤。本发明还进一步提供了一种脂质复合物,由所述阳离子脂质体和带负电的活性物质(待装载/递送目标物)制成。优选的,所述活性物质指具有生物或药理活性的物质,可为核酸分子或蛋白分子;当为所述核酸分子时,所述阳离子脂质体与带负电的核酸分子的n:p比可为20~50:1。所述核酸可以包括至少一个经过修饰的核酸类似物,或是包含核酸衍生物的核苷酸聚合物。所述阳离子脂质体和所述活性物质通过正负电荷静电相互作用形成脂质复合物。该脂质复合物可形成分散在水相中的粒径为80~600nm的纳米颗粒。该纳米颗粒均一且稳定。本发明还提供了一种试剂、试剂盒、制剂或药物组合物,其为包括本发明所述的阳离子脂质分子、阳离子脂质体或脂质复合物。本发明还提供了上述阳离子脂质分子、阳离子脂质体、脂质复合物、试剂、试剂盒、制剂或药物组合物在制备具有下述1)-4)中任一种功能的产品中的应用:1)包封活性物质;2)将活性物质递送至细胞、组织或器官;3)使活性物质在细胞、组织或器官中发挥活性;4)预防、诊断和/或治疗疾病。本发明提供的阳离子脂质分子的合成工艺简单、稳定性好,其阳离子脂质体兼具高效(可体现为高转染效率)和低毒性;同时稳定均一、易于制备;可用于各种细胞系转染。从而具备优秀的传递性,可将活性物质(如示例的sirna)高效递送到细胞(如示例的肺癌细胞)、组织、器官中,实现活性物质的高效调控。解决了现有技术中存在的阳离子脂质体的毒性和传递效率不高的问题。附图说明图1为旋转蒸发法制备的阳离子脂质体以及对应的脂质体/sirna复合物的粒径(图1(a))和表面电位(图1(b))。图2为乳化法制备的阳离子脂质体采用100nm膜过滤前和过滤后的粒径(图2(a))和表面电位(图2(b))。图3为长循环阳离子脂质体采用100nm膜过滤前和过滤后的粒径(图3(a))和表面电位(图3(b))。图4为不同氮磷(n/p)比的脂质体/sirna复合物的粒径(图4(a))和表面电位(图4(b))。图5为脂质体/cy3-sirna复合物转染细胞荧光显微镜图片。图6为脂质体/sirna复合物转染细胞后细胞绿色荧光蛋白的表达水平。图7为空载脂质体细胞活性检测。具体实施方式下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。表3为本发明中英文或缩写的中文参照表,表4所示为各细胞系。表3英文或缩写中文参照表表4细胞系表a549人肺腺癌细胞系mda-mb-231人乳腺癌细胞mcf-7人乳腺癌细胞系h1299非小细胞肺癌细胞实施例1、化合物i-1的制备(1)化合物3的合成在单口瓶中加入油酸1、hbtu(1.1eq)、diea(1.2eq)溶解于二氯甲烷。室温搅拌2h后加入二乙醇胺2(1.2eq)。加毕,室温搅拌18h。反应液分别用1nhcl、饱和na2co3溶液、水洗涤后,加无水硫酸钠干燥。过滤、浓缩滤液至干得粗品。粗品过硅胶柱(石油醚:乙酸乙酯v:v=1:1),制得淡黄色油状物3,收率为58.2%。1hnmr(400m,cdcl3)δppm:(5.305-5.388,m,2h),(3.811-3.836,t,2h),(3.761-3.787,t,2h),(3.531-3.556,t,2h),(3.482-3.508,t,2h),(2.368-2.406,t,2h),(2.005-2.019,m,4h),(1.603-1.638,t,2h),(1.272-1.308,m,20h),(0.885-0.901,t,3h)。esi-ms:m/z392.93[m+na]+,761.74[2m+na]+。(2)化合物5的合成在单口瓶中加入n-羟乙基吗啉4、四溴化碳(1.1eq)溶解于二氯甲烷。然后缓慢加入三苯基膦(1.1eq)。加毕,撤去冰浴,室温搅拌30min。将反应液浓缩至干。加入石油醚500ml,析出大量固体。过滤,将滤液浓缩至干得其粗品。粗品过硅胶柱(石油醚:乙酸乙酯v:v=5:1),制得中间体(2-(4-吗啉)乙基溴),直接用于下一步。另取双口瓶,向其中加入化合物3,无水四氢呋喃,冰浴搅拌下,缓慢加入nah(4.0eq)。加毕,置换氮气,升温至60℃反应1h。将制得的透明油状物用无水四氢呋喃溶解后加入至反应液中。加毕,继续60℃反应16h。加水淬灭反应,用乙酸乙酯萃取,无水硫酸钠干燥。过滤、浓缩滤液至干得粗品。粗品过硅胶柱,制得淡黄色油状物5,收率为37.3%。esi-ms:m/z596.94[m+h]+,618.81[m+na]+,1213.68[2m+na]+。(3)化合物6的合成在单口瓶中加入4.0g化合物5和碘甲烷(10eq)溶于乙腈。升温至70℃反应20h。将反应液浓缩至干,得黄色粗品。取粗品用c18柱分离,制得淡黄色固体6,收率为40.6%。esi-ms:m/z313.08[m-2i-]/2,752.47[m-i-]。1hnmr(400m,cdcl3)δppm:(5.321-5.332,d,2h),(4.066-4.136,m,16h),(3.562-3.799,m,22h),(2.309,s,2h),(1.996-2.003,m,4h),(1.548,s,2h),(1.232-1.298,m,20h),(0.857-0.884,t,3h)。(4)化合物i-1的合成在单口瓶中加入氯型交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物6,无水乙醇,室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌2h。过滤,将滤液浓缩至干,得无水透明固体i-1,收率96.0%。esi-ms:m/z313.12[m-2cl-]/2,660.57[m-cl-]。1hnmr(400m,cdcl3)δppm:(5.366,s,2h),(4.079-4.134,m,16h),(3.552-3.786,m,22h),(2.322,s,2h),(2.037,s,4h),(1.579,s,2h),(1.297-1.324,m,20h),(0.909,s,3h)。实施例2、化合物i-2的制备(1)化合物8的合成在单口瓶中加入芥酸7,搅拌溶解于二氯甲烷,。加入hbtu(1.05eq)和diea(2eq),加毕,室温搅拌2h。加入二乙醇胺(2eq)。加毕,室温搅拌18h。反应液分别用水和稀盐酸洗涤后,用无水硫酸钠干燥。过滤,浓缩滤液至干得粗品。粗品过硅胶柱,制得化合物8,收率约83%。esi-ms:m/z873.22[2m+na]+。(2)化合物9的合成在单口瓶中加入n-羟乙基吗啉4,四溴化碳(1.1eq)和适量二氯甲烷,搅拌溶解后缓慢加入三苯基膦(1.1eq)。加毕,撤去冰浴,室温搅拌30min。将反应液浓缩至干。加入石油醚,析出大量固体。过滤,将滤液浓缩至干得其粗品。粗品过硅胶柱,制得中间体(2-(4-吗啉)乙基溴),直接用于下一步。在双口瓶中加入化合物8和无水四氢呋喃,0℃下,缓慢加入nah(4.0eq)。加毕,置换氮气,升温至60℃反应1h。将上述制得的中间体,用无水四氢呋喃溶解后加入至反应液中。加毕,继续60℃反应18h。加氯化铵淬灭反应,用乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥。过滤,浓缩滤液至干得粗品。粗品过硅胶柱,制得棕色油状物9,收率为47.0%。esi-ms:m/z652.66[m+h]+,1225.50[2m+na]+。(3)化合物10的合成在单口瓶中加入化合物9和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应20h。将反应液浓缩至干,得其粗品。粗品用c18柱分离可制得产物10约,收率为48.8%。esi-ms:m/z341.05[m-2i-]/2,808.36[m-i-]。1hnmr(400m,d2o)δppm:(5.27,s,2h),(3.97-4.02,m,12h),(3.73-3.84,m,6h),(3.54-3.61,m,14h),(3.32,s,3h),(3.30,s,3h),(2.38,s,2h),(1.95-1.97,m,4h),(1.50,s,2h),(1.22-1.24,m,28h),(0.81-0.84,t,3h)。(4)化合物i-2的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物10、无水乙醇,室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,并将滤液浓缩至干,得产物i-2,收率89.5%。esi-ms:m/z341.05[m-2cl-]/2,716.49[m-cl-]。1hnmr(400m,d2o)δppm:(5.26-5.29,m,2h),(3.90-4.70,m,12h),(3.65-3.73,m,6h),(3.47-3.60,m,14h),(3.23,s,6h),(2.31-2.35,m,2h),(1.93-1.96,m,4h),(1.48,s,2h),(1.22-1.25,m,28h),(0.81-0.84,t,3h)。实施例3、化合物i-3的制备(1)化合物11的合成取一单口瓶,向其中加入化合物5和甲醇。待其溶解后加入pd/c,置换氢气后于室温搅拌18h。铺硅藻土过滤,并将滤液浓缩至干,即制得化合物11,直接用于下一步。esi-ms:m/z598.52[m+h]+,620.50[m+na]+。1hnmr(cd3od,ppm):δ(3.69-3.73,m,8h),(3.61-3.66,m,12h),(2.53-2.65,m,12h),(2.45,t,2h),(1.61,t,2h),(1.31-1.35,m,28h),(0.92,t,3h)。(2)化合物12的合成在单口瓶中加入化合物11和dmf,搅拌溶解后加入碘甲烷(10eq)。密闭条件下升温至100℃反应20h。将反应液浓缩至干,用乙酸乙酯洗涤后得其粗品。粗品用c18柱分离,得淡黄色固体12,收率为63.5%。esi-ms:m/z314.17[m-2i-]/2,754.37[m-i-]。1hnmr(400m,d2o)δppm:(3.98-4.05,m,12h),(3.74-3.85,m,6h),(3.55-3.68,m,14h),(3.33,s,3h),(3.31,s,3h),(2.39,s,2h),(1.52,s,2h),(1.24,s,28h),(0.82-0.86,t,3h)。(3)化合物i-3的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物12,无水乙醇,水,室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,用少量无水乙醇洗涤滤饼。将滤液浓缩至干,得无色透明固体(i-3),收率94.0%。esi-ms:m/z313.95[m-2cl-]/2,662.35[m-cl-]。1hnmr(400m,d2o)δppm:(3.92-4.04,m,12h),(3.67-3.74,m,6h),(3.47-3.62,m,14h),(3.27,s,3h),(3.24,s,3h),(2.33-2.36,t,2h),(1.49-1.51,m,2h),(1.24,s,28h),(0.83-0.86,t,3h)。实施例4、化合物ⅱ-1的制备(1)化合物14的合成在双口瓶中加入化合物13及三乙胺(1.1eq)在thf中搅拌溶解,于0℃下加入mscl(1.05eq)。保持0℃反应1h。加硅藻土过滤,并用少量thf洗涤滤饼,得中间体滤液待用。另取一双口瓶,化合物3溶解于thf后,0℃下加入nah(3eq)。加毕,置换氮气,升温至60℃反应1h。用注射器加入上述中间体滤液(约3eq)。加毕,继续60℃反应2h。加水淬灭反应,用乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥。过滤,浓缩滤液至干得粗品。粗品经柱层析分离,制得化合物14,直接用于下一步。esi-ms:m/z1125.20[2m+na]+。(2)化合物15的合成在单口瓶中加入化合物14和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应20h。将反应液浓缩至干,得其粗品。粗品经c18柱层析分离,制得产物15。esi-ms:m/z290.95[m-2i-]/2,708.17[m-i-]。1hnmr(400m,d2o)δppm:(8.76-8.78,d,2h),(8.69-8.71,d,2h),(7.91-7.93,d,4h),(5.22-5.24,m,2h),(4.87,s,2h),(4.80,s,2h),(4.30,s,3h),(4.28,s,3h),(3.63-3.81,m,8h),(2.42-2.45,m,2h),(1.89-1.91,m,4h),(1.51,s,2h),(1.16-1.21,m,20h),(0.76-0.79,m,3h)。(3)化合物ⅱ-1的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物15,无水乙醇,室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,并将滤液浓缩至干,得产物(ⅱ-1),收率约90.9%。esi-ms:m/z290.91[m-2cl-]/2,615.99[m-cl-]。1hnmr(400m,d2o)δppm:(8.69-8.76,m,4h),(7.89-7.91,m,4h),(5.23-5.24,m,2h),(4.70-4.85,m,2h),(4.32,s,3h),(4.30,s,3h),(3.64-3.81,m,8h),(2.42-2.46,m,2h),(1.90-1.91,m,4h),(1.51-1.54,m,2h),(1.17-1.22,m,20h),(0.77-0.80,t,3h)。实施例5、化合物ⅱ-2的制备(1)化合物16的合成在双口瓶中加入化合物13和三乙胺(1.1eq)在thf中搅拌溶解,于0℃下加入mscl(1.05eq)。维持0℃反应1h。加硅藻土过滤,并用少量thf洗涤滤饼,得中间体滤液直接用于下一步。在双口瓶中加入化合物8,无水四氢呋喃,冰浴搅拌下,缓慢加入nah(3.0eq)。加毕,置换氮气,升温至60℃反应1h。用注射器加入上述中间体滤液。加毕,继续60℃反应2h。加水淬灭反应,用乙酸乙酯萃取,无水硫酸钠干燥。过滤,浓缩滤液至干得粗品。粗品经柱层析分离过柱可制得化合物16,收率为53.9%。esi-ms:m/z1237.33[2m+na]+。1hnmr(cdcl3,ppm):δ(8.35-8.60,m,4h),(7.19-7.28,m,4h),(5.34-5.36,m,2h),(4.50-4.63,m,4h),(3.61-3.78,m,8h),(2.38-2.42,m,2h),(1.99-2.04,m,4h),(1.62-1.65,m,2h),(1.27-1.35,m,28h),(0.88,t,3h)。(2)化合物17的合成在单口瓶中加入化合物16和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应20h。将反应液浓缩至干,用石油醚洗涤得其粗品。粗品用c18柱层析分离,制得淡黄色产物17,收率为31.1%。esi-ms:m/z318.96[m-2i-]/2,764.21[m-i-]。1hnmr(d2o,ppm):δ(8.81-8.83,d,2h),(8.72-8.74,d,2h),(7.94-7.97,dd,4h),(5.26,s,2h),(4.90,s,2h),(4.83,s,2h),(4.34,s,3h),(4.31,s,3h),(3.66-3.85,m,8h),(2.46,s,2h),(1.92-1.96,m,4h),(1.54,s,2h),(1.21-1.28,m,28h),(0.82,t,3h)。(3)化合物ⅱ-2的合成在单口瓶中加入阴离子交换树脂,用无水甲醇洗涤杂质,除去甲醇,备用。另取一单口瓶,向其中加入化合物17和无水甲醇,室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,并将滤液浓缩至干,得淡棕色产物ⅱ-2,收率94.3%。esi-ms:m/z318.97[m-2cl-]/2,672.08[m-cl-]。1hnmr(d2o,ppm):δ(8.69-8.78,m,4h),(7.89-7.92,dd,4h),(5.23-5.26,m,2h),(4.85,s,2h),(4.79,s,2h),(4.33,s,3h),(4.30,s,3h),(3.63-3.82,m,8h),(2.44,t,2h),(1.90-1.93,m,4h),(1.53,s,2h),(1.19-1.22,m,28h),(0.80,t,3h)。实施例6、化合物ⅲ-1的制备(1)化合物20的合成在单口瓶中加入化合物18、dmf和二氯甲烷,搅拌悬浮。0℃下加入草酰氯(4.5eq),加毕,升温至50℃反应1h,溶清。继续于50℃下反应3h。将反应液浓缩至干得黄色固体,加入二氯甲烷得其酰氯溶液,备用。另取一单口瓶,加入化合物19(2.1eq)、无水吡啶和二氯甲烷,搅拌溶解。0℃下滴入上述酰氯溶液。加毕,室温反应2h,搅拌过程中有大量固体析出。倒出母液,得残留粗品。粗品经柱层析分离,制得血红色粘稠油状物20,直接用于下一步。esi-ms:m/z464.48[m+h]+。(2)化合物21的合成在双口瓶中加入化合物20在甲醇中搅拌溶解。加入pd/c,两瓶口分别套上氢气球,置换氢气。室温反应16h,重新更换两个氢气球。继续室温反应至反应完全。铺硅藻土过滤,浓缩滤液至干得粗品。粗品经柱层析分离,制得淡棕色粘稠油状物21,收率95.2%。esi-ms:m/z434.44[m+h]+,889.47[2m+na]+。1hnmr(400m,dmso-d6)δppm:(8.447,s,2h),(7.464,s,1h),(7.110,s,2h),(5.417,s,2h),(3.256-3.271,m,8h),(3.168,s,4h),(2.419,s,12h),(1.682-1.715,m,4h)。(3)化合物22的合成在单口瓶中加入化合物1、dmf和二氯甲烷。搅拌溶解后于0℃下加入草酰氯(2.5eq)。加毕,升温至50℃回流2h。将反应液浓缩至干,加入二氯甲烷得其酰氯溶液,备用。另取一单口瓶,加入化合物21(0.8eq)、无水吡啶(3.0eq)和dmf。搅拌溶解后于冰浴下滴入上述酰氯溶液。加毕,于室温反应2h。浓缩除去溶剂得粗品,粗品经柱层析分离,制得产物22,收率65.6%。esi-ms:m/z698.78,[m+h]+,1395.66,[2m+h]+。1hnmr(cdcl3,ppm):δ(9.314,s,1h),(8.357,s,2h),(8.165,s,2h),(8.018,s,1h),(5.313-5.356,m,2h),(3.817,s,8h),(3.482,s,4h),(2.946,s,12h),(2.433,s,2h),(2.002-2.016,m,8h),(1.674,s,2h),(1.270-1.304,m,20h),(0.891,t,3h)。(4)化合物23的合成在单口瓶中加入化合物22和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应24h。将反应液浓缩至干,用石油醚-乙酸乙酯体系洗涤得其粗品。粗品经c18柱分离,制得淡黄色固体23,收率为21.3%。esi-ms:m/z364.60[m-2i-]/2,854.59[m-i-]。1hnmr(cdcl3,ppm):δ(8.090,s,2h),(7.943,s,1h),(5.335,s,2h),(4.016,s,8h),(3.513-3.644,m,16h),(3.232,s,6h),(2.454,s,2h),(2.152,s,4h),(2.007,s,4h),(1.641,s,2h),(1.273,s,20h),(0.883,s,3h)。(5)化合物ⅲ-1的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物23,无水乙醇和水室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌20h。过滤,并将滤液浓缩至干,得淡黄色固体ⅲ-1,收率约88%。esi-ms:m/z364.85,[m-2cl-]/2,763.19,[m-cl-]。1hnmr(cdcl3,ppm):δ(7.97,s,2h),(7.85,s,1h),(5.23-5.24,d,2h),(3.91,t,8h),(3.39-3.51,m,16h),(3.12,s,6h),(2.33,s,2h),(2.05,s,4h),(1.91,s,4h),(1.54,s,2h),(1.17-1.20,m,20h),(0.79,t,3h)。实施例7、化合物ⅲ-2的制备(1)化合物24的合成取一单口瓶,向其中加入化合物22,加入甲醇搅拌。待其溶解后加入pd/c,置换氢气后于室温搅拌18h。铺硅藻土过滤,并将滤液浓缩至干,即制得固体化合物24,收率90.6%。esi-ms:m/z700.55[m+h]+。(2)化合物25的合成在单口瓶中加入化合物24和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应24h。将反应液浓缩至干,用石油醚-乙酸乙酯体系洗涤得其粗品。取一半粗品经c18柱层析分离,制得产物25,收率为25.8%。esi-ms:m/z364.85[m-2i-]/2,856.48[m-i-]。1hnmr(d2o,ppm):δ(8.00,s,2h),(7.86,s,1h),(3.93,s,8h),(3.43-3.57,m,16h),(3.15,s,6h),(2.37,s,2h),(2.07,s,4h),(1.56,s,2h),(1.21,s,28h),(0.82,s,3h)。(3)化合物ⅲ-2的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物25,无水乙醇,水室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,并将滤液浓缩至干,得产物ⅲ-2,收率约92%。esi-ms:m/z364.94,[m-2cl-]/2,764.40[m-cl-]。1hnmr(d2o,ppm):δ(7.96,s,2h),(7.85,s,1h),(3.89-3.92,t,8h),(3.39-3.49,m,16h),(3.12,s,6h),(2.34,s,2h),(2.04,s,4h),(1.54,s,2h),(1.19,s,28h),(0.80,t,3h)。实施例8、化合物ⅳ-1的制备(1)化合物27的合成在单口瓶中加入化合物1,dmf和二氯甲烷。0℃条件下加入氯化亚砜(2.0eq),加毕,升温至室温反应2h。将反应液浓缩至干,加入二氯甲烷得其酰氯溶液,备用。另取一单口瓶,加入化合物26(0.73eq)、三乙胺和二氯甲烷。冰浴搅拌下滴入上述酰氯溶液。加毕,室温反应1h。分别用水、稀氢氧化钠和稀盐酸洗涤。无水硫酸钠干燥。过滤,浓缩滤液至干得粗品。粗品经柱层析分离,制得产物27,收率73.5%。esi-ms:m/z969.15[2m+na]+。(2)化合物28的合成在单口瓶中加入化合物27,四氢呋喃和水。搅拌下加入无水氢氧化锂(4.0eq),加毕,室温反应18h。浓缩除去四氢呋喃,残留物用4nhcl调节ph至2,析出大量固体。过滤,滤饼浓缩至干,制得类白色固体产物28。esi-ms:m/z889.74[2m-h]-。(3)化合物30的合成在单口瓶中加入化合物28,hbtu(2.2eq),diea(3.0eq)和dmf。室温搅拌1h。加入化合物29(2.2eq)。加毕,室温搅拌2h。加水淬灭反应,并用二氯甲烷萃取。有机相用水洗涤3次,加无水硫酸钠干燥。过滤、浓缩滤液至干得粗品。粗品用二氯甲烷—甲醇体系过柱,制得淡橙色固体30,收率约92%。esi-ms:m/z1173.33[2m+na]+。(4)化合物31的合成在单口瓶中加入化合物30和无水甲醇,搅拌下加入dmf和碘甲烷。密闭升温至60℃反应24h。将反应液浓缩至干,用石油醚-乙酸乙酯体系洗涤得其粗品。粗品经c18柱层析分离,制得淡黄色固体产物31,收率为24.8%。esi-ms:m/z327.95[m-2i-]/2,782.17[m-i-]。1hnmr(d2o,ppm):δ(8.56-8.58,t,4h),(8.03,s,2h),(7.95,s,1h),(7.84-7.85,d,4h),(5.24,s,2h),(4.64,s,4h),(4.17,s,6h),(2.38,s,2h),(1.92,s,4h),(1.56,s,2h),(1.18-1.21,m,20h),(0.78-0.79,t,3h)。(5)化合物ⅳ-1的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物31,无水乙醇,水室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,并将滤液浓缩至干,得产物ⅳ-1,收率约93%。esi-ms:m/z327.94[m-2cl-]/2,690.23[m-cl-]。1hnmr(d2o,ppm):δ(8.56-8.58,m,4h),(7.81-8.02,m,7h),(5.22,s,2h),(4.66-4.71,m,4h),(4.18-4.20,m,6h),(2.34,s,2h),(1.90,s,4h),(1.55,s,2h),(1.16-1.23,m,20h),(0.76-0.79,t,3h)。实施例9、化合物ⅴ-1的制备(1)化合物33的合成在单口瓶中加入化合物32、dmf和二氯甲烷,搅拌溶解。0℃下加入草酰氯(2.0eq),加毕,升温至50℃回流2h。将反应液浓缩至干,加入二氯甲烷得其酰氯溶液,备用。另取一个单口瓶,加入化合物21(0.95eq)、无水吡啶(3.0eq)、dmf,搅拌溶解。零度下滴入上述酰氯溶液。加毕,室温反应2h。浓缩除去溶剂,用硅胶柱层析分离,得淡黄色油状物33,收率71.7%。esi-ms:m/z576.64[m+h]+,1151.60[2m+h]+,1173.55[2m+na]+。(2)化合物34的合成在单口瓶中加入化合物33、四氢呋喃和水,搅拌溶解。加入无水氢氧化锂(4.0eq),加毕,室温反应20h。浓缩除去四氢呋喃,补加水,用二氯甲烷洗涤,水相用6nhcl调节ph至6。将水相浓缩至干,制得含无机盐的油状粗品34,直接用于下一步。esi-ms:m/z562.51[m+h]+,568.59[m+li]+。(3)化合物36的合成称取化合物35于一单口瓶中,加入二氯甲烷溶解,待用。另取一单口瓶,向其中加入化合物34和无水dmf,搅拌下加入hobt(1.5eq)、diea(3eq)和化合物35的二氯甲烷溶液。加毕,室温搅拌半小时。再加入hbtu(1.5eq),加毕,室温反应16h。将反应液浓缩至干,加入二氯甲烷溶解残留物,用水洗涤后加入无水硫酸钠干燥。过滤,并将滤液浓缩至干。过硅胶柱,得淡黄色油状物36,收率47.7%。esi-ms:m/z1106.14[m+h]+,1128.07[m+na]+。1hnmr(400m,cdcl3)δppm:(9.581,s,1h),(8.294-8.297,d,2h),(8.033-8.058,t,2h),(7.969,s,1h),(6.948,s,1h),(5.305-5.326,m,1h),(3.681-3.704,t,8h),(3.616-3.645,m,12h),(3.512-3.562,m,6h),(3.442-3.469,t,2h),(3.106-3.211,m,1h),(2.483-2.541,m,14h),(2.248-2.385,m,4h),(1.725-2.015,m,12h),(0.995-1.582,m,22h),(0.970,s,3h),(0.898-0.915,d,3h),(0.850-0.871,dd,6h),(0.664,s,3h)。(4)化合物37的合成在单口瓶中加入化合物36和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应24h。补加碘甲烷,继续100℃密闭反应24h。将反应液浓缩至干,用石油醚/乙酸乙酯体系洗涤得其粗品。粗品过c18柱,得淡黄色固体23,收率为25.5%。esi-ms:m/z568.36[m-2i-]/2,1261.99[m-i-]。(5)化合物ⅴ-1的合成在单口瓶中加入阴离子交换树脂,用无水乙醇洗涤杂质,除去乙醇,备用。另取一单口瓶,向其中加入化合物37,无水乙醇,室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,用少量无水乙醇洗涤滤饼。将滤液浓缩至干,得无色透明固体18,收率98.0%。esi-ms:m/z568.17[m-2cl-]/2,1169.87[m-cl-]。1hnmr(400m,d2o)δppm:(7.93,s,2h),(7.85,s,1h),(5.47,s,1h),(4.01,s,8h),(3.37-3.69,m,34h),(3.21,s,6h),(0.73-2.36,m,54h)。实施例10、化合物ⅵ-1的制备(1)化合物40的合成在单口瓶中加入苯甲醇38和三乙胺(1.1eq)搅拌溶解于二氯甲烷。称量氯甲酸苯酯39(1.0eq),于0℃下缓慢滴入。加毕,室温搅拌18h。反应液分别用水和稀盐酸洗涤,并用nahco3调ph至8。加无水硫酸钠干燥有机相。过滤,将滤液浓缩至干,得其粗品。粗品过硅胶柱,制得透明油状物40,收率约为82%。1hnmr(400m,cdcl3)δppm:(7.38-7.49,m,7h),(7.20-7.32,m,3h),(5.30,s,2h)。(2)化合物42的合成在单口瓶中加入化合物40和二氯甲烷,搅拌溶解。称取化合物41(1.0eq),滴入至反应液中,加毕,于室温反应40h。反应液分别用水和稀氢氧化钠洗涤,将有机相浓缩至近干,加盐酸成盐并用乙醇重结晶,制得白色固体42的盐酸盐。用碳酸钠游离,制得类白色固体42,收率为53.0%。esi-ms:m/z372.09[m+h]+。1hnmr(400m,cd3od)δppm:(7.34-7.37,m,10h),(5.12,s,4h),(3.45-3.48,m,4h),(3.18-3.21,m,4h)。(3)化合物43的合成在单口瓶中加入化合物32(1.0eq)、hbtu(1.1eq)、diea(2.0eq)于二氯甲烷中搅拌溶解。室温搅拌反应1h。加入化合物42(0.95eq),加毕室温搅拌反应20h。反应液分别用水、稀盐酸和碳酸氢钠洗涤,加入无水硫酸钠干燥。过滤,浓缩滤液至干,得粗品。粗品过硅胶柱,得油状物43,收率为91.6%。1hnmr(cdcl3,ppm):δ(7.34-7.36,m,10h),(5.08-5.12,d,4h),(3.66,s,3h),(3.29-3.48,m,8h),(2.29-2.37,m,4h),(1.60-1.65,m,4h)。(4)化合物44的合成在单口瓶中加入化合物43于四氢呋喃中搅拌溶解。称取无水氢氧化锂(4.0eq)于水中溶解后加入至上述反应液中,加毕,室温反应6h。浓缩除去四氢呋喃,用石油醚-乙酸乙酯体系洗涤两次,水相用4nhcl调节ph至2。用二氯甲烷萃取,无水硫酸钠干燥。过滤,滤液浓缩至干得化合物8,收率约82.3%。esi-ms:m/z498.09[m-h]-。(5)化合物45的合成在单口瓶中加入化合物44(1.0eq),无水dmf,二氯甲烷,搅拌溶解。缓慢加入氯化亚砜(2.0eq),加毕,升温至40℃回流2h。将反应液浓缩至干,加入二氯甲烷得其酰氯溶液,备用。另取一个单口瓶,加入化合物26(0.9eq)和三乙胺(2.0eq)于二氯甲烷中搅拌溶解。0℃下滴入上述酰氯溶液。加毕,室温反应2h。反应液分别用1m氢氧化钠、1m盐酸及饱和碳酸氢钠洗涤,无水硫酸钠干燥。过滤,浓缩滤液至干,得粗品。粗品过硅胶柱,得类白色固体45,收率为80.6%。esi-ms:m/z713.32[m+na]+,1402.80[2m+na]+。1hnmr(400m,cdcl3)δppm:(8.48-8.49,d,2h),(8.31-8.32,t,1h),(7.27-7.33,m,10h),(5.04-5.08,d,4h),(3.93,s,6h),(3.42-3.47,m,4h),(3.27-3.33,m,4h),(2.37-2.42,m,4h),(1.64-1.70,m,4h)。(6)化合物46的合成在双口瓶中加入化合物45、甲醇和乙酸乙酯,搅拌溶解。加入pd/c后,两个瓶口均套上氢气球,置换氢气。室温反应16h,重新更换两个氢气球。继续室温反应4h,铺硅藻土过滤,浓缩滤液至干,得透明油状物46,收率70.3%。esi-ms:m/z421.38[m-h]-。(7)化合物47的合成在单口瓶中加入化合物1、dmf、二氯甲烷,搅拌溶解。冰浴下加入氯化亚砜(2.0eq),加毕,升温至40℃反应2h。将反应液浓缩至干得,加入二氯甲烷得其酰氯溶液,备用。另取一单口瓶,加入化合物46(0.45eq)、三乙胺(2.0eq)溶解于二氯甲烷。冰浴搅拌下滴入上述酰氯溶液。加毕,室温反应1h。反应液分别用水、稀氢氧化钠、稀盐酸和饱和氯化钠溶液洗涤后加无水硫酸钠干燥。过滤,滤液浓缩至干。二氯甲烷溶解后进行柱层析分离,制得产物约17.4g。esi-ms:m/z973.77[m+na]+,1hnmr(cd3od,ppm):δ(8.51-8.52,d,2h),(8.34,t,1h),(5.29-5.37,m,4h),(3.95,s,6h),(3.48-3.51,m,4h),(3.33-3.39,m,4h),(2.45-2.50,m,4h),(2.14-2.22,m,4h),(1.99-2.05,m,8h),(1.71-1.78,m,4h),(1.55-1.61,m,4h),(1.27-1.33,m,40h),(0.89-0.92,m,6h)。(8)化合物48的合成在单口瓶中加入化合物47溶解于四氢呋喃。称取无水氢氧化锂(8.0eq),用水溶解后加入至上述反应液中,加毕,室温反应18h。浓缩除去四氢呋喃,水相用4nhcl调节ph至2。用二氯甲烷萃取,无水硫酸钠干燥。过滤,滤液浓缩至干得产物48,收率约95.0%。esi-ms:m/z922.03[m-h]-。(9)化合物49的合成在单口瓶中加入化合物48,hbtu(2.4eq),diea(3.0eq)溶解于二氯甲烷,室温搅拌1h。加入化合物16(2.4eq)。加毕,室温搅拌3h。反应液分别用水、1n氢氧化钠洗涤后,加无水硫酸钠干燥。过滤,浓缩滤液至干得粗品。粗品用二氯甲烷—甲醇体系柱层析,制得淡黄色油状物49,收率46.6%。esi-ms:m/z1175.92[m+h]+。1hnmr(cdcl3,ppm):δ(10.59,d,1h),(8.70,s,2h),(7.94-8.07,m,4h),(7.53,s,1h),(5.31-5.38,m,4h),(3.70-3.72,m,8h),(3.49-3.63,m,12h),(2.41-2.56,m,15h),(2.22-2.26,m,2h),(1.95-2.10,m,11h),(1.48-1.84,m,12h),(1.08-1.36,m,40h),(0.87-0.91,m,6h)。(10)化合物50的合成在单口瓶中加入化合物49和无水dmf,搅拌下加入碘甲烷。密闭升温至100℃反应18h。将反应液浓缩至干,用石油醚洗涤得其粗品。粗品用c18柱层析分离,制得淡黄色固体产物50,收率为50.2%。esi-ms:m/z602.67[m-2i-]/2,1331.73[m-i-]。1hnmr(d2o,ppm):δ(8.29-8.30,d,2h),(8.21-8.22,m,1h),(5.34-5.37,m,4h),(4.02-4.05,m,8h),(3.68-3.71,m,4h),(3.48-3.66,m,16h),(3.34-3.40,m,4h),(3.28,s,6h),(2.47-2.52,m,4h),(2.15-2.26,m,8h),(2.01-2.08,m,8h),(1.70-1.81,m,4h),(1.59-1.63,m,4h),(1.30-1.34,m,40h),(0.90-0.94,m,6h)。(11)化合物ⅵ-1的合成在单口瓶中加入阴离子交换树脂,用乙腈洗涤杂质,除去乙腈,备用。另取一单口瓶,向其中加入化合物50,乙腈,水室温搅拌。待其溶解后加入至上述树脂中,加毕,于室温搅拌18h。过滤,用少量乙腈洗涤滤饼。将滤液浓缩至干,并加少量甲苯旋干得产物ⅵ-1,收率约73%。esi-ms:m/z602.66[m-2cl-]/2,1239.86[m-cl-]。1hnmr(d2o,ppm):δ(8.29-8.29,d,2h),(8.23-8.23,t,1h),(5.34-5.37,m,4h),(4.02-4.04,m,8h),(3.62-3.67,m,4h),(3.47-3.60,m,16h),(3.34-3.39,m,4h),(3.26,s,6h),(2.47-2.51,m,4h),(2.15-2.24,m,8h),(2.01-2.10,m,8h),(1.70-1.81,m,4h),(1.57-1.63,m,4h),(1.30-1.41,m,40h),(0.90-0.94,m,6h)。实施例11、将本发明合成的i-3、ⅲ-1、ⅲ-2脂质体阳离子化合物和dope(摩尔比:1:1)溶解在氯仿/甲醇混合体系中(10ml,1:1,v/v),倒入旋转蒸发仪的圆底烧瓶中,在50℃水浴恒温,在真空条件下蒸发2h。然后在真空干燥箱40℃真空干燥4h,完全去除有机溶剂。加入生理盐水(6ml),常压,50℃水浴恒温震荡1h得到白色乳液,然后把白色乳液转移到小玻璃瓶中,超声30min至乳液基本透明。最后乳液进行220μm滤器(milliporecorp.)过滤头除菌,转移至新的样品瓶,于4℃保存。使用zetasizernano粒径仪进行粒径、多分散性和表面电荷的测定(图1)。实施例12、用无水乙醇分别配制浓度为0.01mol/l的阳离子化合物ⅰ-1、ⅰ-2、ⅰ-3、ⅱ-1、ⅱ-2、ⅲ-1、ⅲ-2、ⅵ-1、ⅵ-2、ⅴ-1和dope溶液。将阳离子化合物和dope等摩尔比的(50%:50%,mol/mol)乙醇混合液加入ph=5的0.2m醋酸钠缓冲水溶液,涡旋均匀,通风磁力搅拌至混合体系中乙醇完成挥发,补加等量蒸馏水,得到透明乳液。取部分乳液用脂质体挤出仪(mini-extruder,avantilipidspolar,inc,usa)采用100nm过滤膜过滤,使用zetasizernano粒径仪进行粒径对比过滤前后的粒径和表面电位变化(图2)。实施例13、阳离子化合物ⅰ-1、dope(二油酰基磷脂酰乙醇胺)和dspe-peg(二硬脂酰基磷脂酰乙醇胺-聚乙二醇)分别按照摩尔比45%:45%;5%和40%:40%:10%配置乙醇溶液,然后加入ph=5的0.2m醋酸钠缓冲水溶液,涡旋均匀,通风磁力搅拌至混合体系中乙醇完成挥发,补加等量蒸馏水,得到透明乳液。取部分乳液用脂质体挤出仪100nm过滤膜过滤,使用zetasizernano粒径仪进行粒径对比过滤前后的粒径和表面电位变化(图3)。由实施例11、12和13可知,本发明所合成的阳离子化合物可以通过旋转蒸发法和乳化法制备得到无菌的阳离子脂质体。事实上,各种常规制备脂质体的方法,如超声法、反相蒸发法、注入法、冷冻干燥法和高压均质发等,均适合本发明所述阳离子脂质分子制备纳米脂质体。图1中所示是使用zetasizernano粒径仪对旋转蒸发法制备的相关阳离子脂质体粒径和表面电位测定结果。所得的脂质体粒径范围在89~130nm,表面电位范围为30-45mv左右。图2所示为乳化法制备的阳离子脂质体采用脂质体挤出仪100nm膜过滤前后粒径和表面电位测定结果。直接用乳化法制备的脂质体,未采用脂质体挤出仪100nm膜过滤之前,其粒径在100-200nm不等,表面电荷在30~70mv范围。通过脂质体挤出仪100nm膜过滤后,本发明的所有脂质体粒径变的更加均一,在80~120nm范围,而且其表面电位也相应降低,在5~45mv范围,个别样品存在急剧减少现象。图3所示为在ⅰ-1和dope为主要成分的脂质体中添加辅助脂质分子dspe-peg挤出仪(100nm膜)过滤前后粒径和表面电位测定结果。dspe-peg的加入,可以增加脂质体表面亲水性,增加体内循环时间。dspe-peg的增加,脂质体粒径稍微增大(图3(a))。但是其表面电位随着dspe-peg含量的增加而降低,从53.6±1.6mv降至10%dspe-peg的20.0±0.6mv(图3(b))。从上述结果可知,本发明采用不同脂质体的制备方法,添加不一样的辅助脂质分子,均可以得到粒径均匀稳定的纳米脂质体溶液,而且表面电位均为正,证明脂质体表面带有净电荷。实施例14、ⅰ-1/dope脂质体和sirna复合物的制备和表征,具体方法是:取10ul样品稀释在1ml的超纯水后,采用zetasizernano进行粒径和表面电荷的测定。分别取实施举例12制备的ⅰ-1/dope、ⅰ-3/dope、ⅲ-1/dope、ⅲ-2/dope脂质体溶液5μl加入20μlopti-mem培养液,混匀,为溶液1。将1μlsirna(浓度20μm)加入24μlopti-mem培养液,为溶液2。将溶液2滴加到溶液1,混匀,室温静止30min,得到ⅰ-1/dope、ⅰ-3/dope、ⅲ-1/dope、ⅲ-2/dope脂质体和sirna的纳米复合物。然后使用zetasizernano粒径仪进行纳米复合物粒径、多分散性和表面电位的测定(ⅰ-3/dope、ⅲ-1/dope、ⅲ-2/dope脂质体和sirna的纳米复合物测定结果见图1,ⅰ-1/dope脂质体和sirna的纳米复合物测定结果见图4。)图4所示为不同氮磷(n/p)比的ⅰ-1脂质体和sirna复合物的粒径和表面电位测试结果。阳离子脂质体和sirna核酸通过静电作用形成复合纳米粒子。从图4上可以观察到,随着n/p比的增加,复合物表面电荷随之升高。当阳离子脂质体ⅰ-1与sirna的n/p比为10:1时,其粒径为581±44nm,表面电位为-18.7±5.6mv,是负值。说明在此n/p比,脂质体不能很好的对sirna进行压缩,形成良好的复合物。当n/p比为20:1时,其粒径为218±61nm,表面电位为13.3±2.4mv。脂质体与sirna复合物表面所携带的电荷量是影响转染效率的一个重要因素。表面电位是用来衡量材料之间引力或斥力强度的物理量,表面电位过低,纳米粒子越易于凝聚,不但会影响体系的稳定性,而且其包裹核酸分子的能力也较弱,进而导致转染效率下降。而且由于细胞膜带负电荷,载体材料与核酸所形成的复合物呈现正电性,可以帮助纳米复合物更加容易进入细胞内部。因此,对于脂质体sirna复合物来说,合适的表面电位将有助于保证复合物有较高的转染效率,以及在体液运输中稳定存在。由本实施例14可以看出,本发明的阳离子脂质体可以通过正负电荷相互作用,跟核酸自发形成脂质体/核酸复合物。事实上,各种常规制备脂质体-核酸复合物的方法,如脂质体薄膜和核酸溶液水合发、冻干-再水合法等方法,均适合于本发明所述的脂质体-核酸复合物的制备。同样,本发明的阳离子脂质体还合适除sirna之外的各种核酸包裹复合实验,这些核酸包括但是并不局限于dna、信使mrna、microrna等,都能有效的形成颗粒小,均一稳定脂质体/核酸复合物。实施例15、ⅰ-1、ⅰ-3、ⅲ-1、ⅲ-2、ⅴ-1、ⅵ-1和ⅵ-2脂质体与cy3荧光标记sirna(cy3-sirna)复合物转染a549细胞。将a549细胞以2×105个细胞/孔的密度铺到24孔板上,加入含有10%小牛血清的dmem培养液培养24小时至细胞密度达到70%左右。移除培养液,用pbs洗两遍。将实施例14制备的脂质体/sirna复合物稀释至500μlopti-mem培养液,加入24孔板中。37℃、5%co2条件下培养箱孵育4h。移除含有脂质体/sirna复合物的培养液,加入含有10%小牛血清的dmem培养液孵育24小时。然后采用荧光显微镜观察cy3-sirna的转染情况,激发光550nm,发射光570nm。图5所示为脂质体/cy3-sirna复合物的转染结果荧光显微镜图片,红色荧光为被脂质体转染进入细胞的cy3-sirna。市面上出售的lipofatamine2000(lipo2000)转染试剂可以把cy3-sirna传递到细胞里面,本发明的脂质体同样可以把cy3-sirna转染进细胞质。而且对比,本发明某些脂质体转染的细胞其红色荧光强度大大强于lipo2000,说明本发明的某些脂质体可以更加高效的转染cy3-sirna,比如ⅲ-1、ⅴ-1和ⅵ-1脂质体。实施例16、将表达绿色荧光蛋白(gfp)的a549细胞以2×105个细胞/孔的密度铺到24孔板上,加入含有10%小牛血清的dmem培养液培养24h至细胞密度达到70%左右。移除培养液,用pbs洗两遍。将实施举例14的方法制备的脂质体/sirna复合物稀释至500μlopti-mem培养液,加入24孔板中。37℃、5%co2条件下培养箱孵育4h。移除含有脂质体/sirna复合物的培养液,加入含有10%小牛血清的dmem培养液孵育48h。移除培养液,用pbs洗两遍,用胰酶将细胞消化下来,4℃,1000rpm离心5min得到细胞沉淀,用pbs洗涤细胞沉淀,4℃,1000rpm离心5min,弃上清。反复洗涤两次,将细胞沉淀重新悬浮在无血清dmem培养基,使用流式细胞仪(bd,facscalibur,bdbiosciences),检测每1×104细胞的相对荧光强度。由实施例16可以出来,通过实施例15和实施例16类似的细胞转染操作,本发明所述的新型阳离子脂质体/sirna复合物均可以实现将sirna传递进入包括但是不局限于a549、mda-mb-231、mcf-7细胞等各类细胞。事实上,对于某些特定的细胞,本发明所述的阳离子脂质体组合库中会有特定的脂质体可以高效的传递sirna进入这些细胞。图6所示为举例的脂质体/sirna复合物转染细胞后细胞绿色荧光蛋白的表达水平。本发明所述的脂质体制备得到的脂质体/sirna复合物对细胞进行转染,能够将sirna对应基因的表达有效的降低。随着调整脂质体和sirna的n/p比例,本发明的脂质体/sirna复合物对目标基因的下调相当于甚至优于市面上的转染试剂标杆试剂lipo2000。实施例17、将a549细胞接种于96孔板(约1.5×104个细胞/孔),每孔加入100μl新配细胞悬液(含10%fbs的培养基),在37℃,5%co2的环境中孵育。24小时后,弃去培养基,并用pbs清洗两次,每孔加入100μl本发明实施例12制备的脂质体颗粒,每个样品设置3个平行组。细胞在37℃下再次孵育24小时后,补加20μlcck-8试剂,再于培养箱中培养2h,通过酶标仪读取在450nm波长处的吸光度进行细胞毒理检测。以未刺激细胞在450nm波长的吸光度作为细胞存活率100%,表示各种阳离子脂质体刺激下的细胞存活率。由实施例17可以看出,通过进行和实施例17相似的细胞毒性检测操作,均可以实施本发明所述空载阳离子脂质体对包括但不局限于a549、mda-mb-231、mcf-7和h1299细胞等各种细胞系的毒性进行检测和评价。图7表示的是本发明的空载各种阳离子脂质体对a549细胞毒性的甲醇。在同等用量下,列举的空载阳离子对细胞系具有非常小的毒性,有的脂质体甚至可以对细胞生长存在促进作用。对比市场上的lipo2000转染试剂,本发明的阳离子脂质体显示出更良好的生物相容性。虽然本发明已将较佳实施例描述如上,然其并非用以限定本发明的内容,任何熟悉本领域的研究人员,在不脱离本发明的主要精神和内容范围内,可以做各种更动和润饰,因此本发明的保护范围应以申请专利的实际要求范围为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1