基因缺失突变的荧光检测试剂盒和荧光检测方法与流程
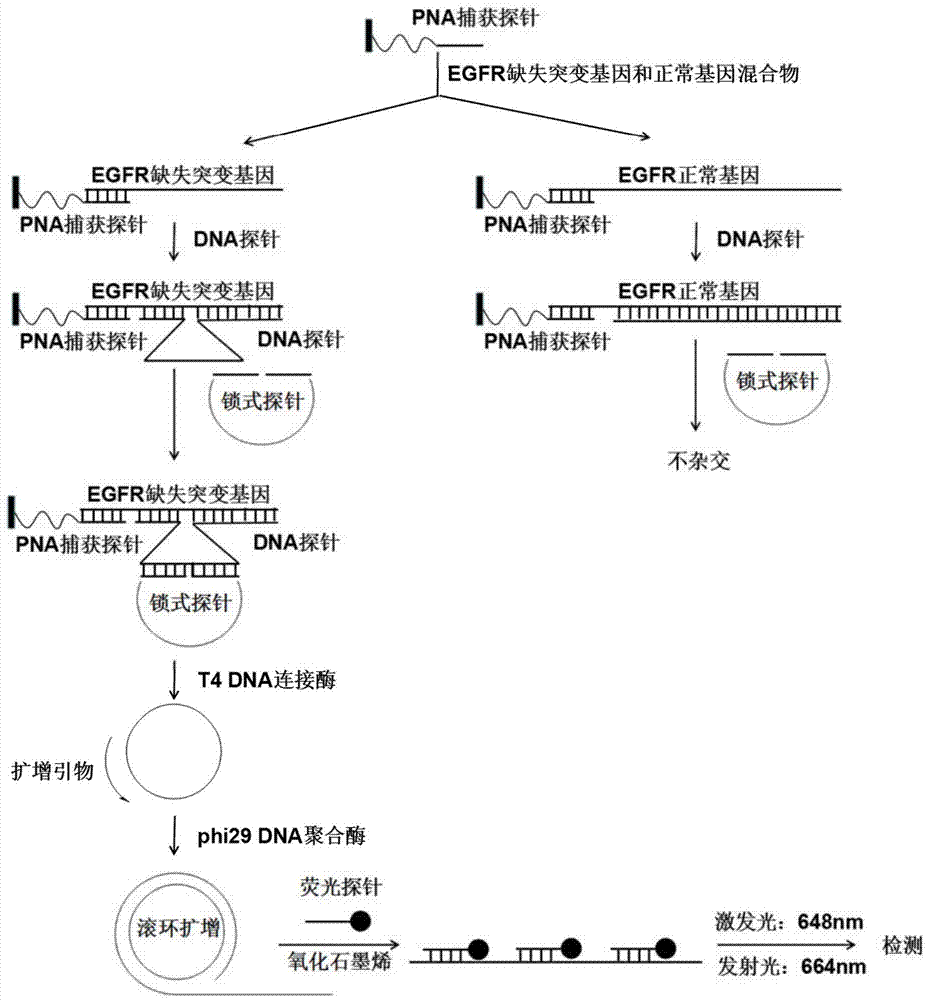
本申请涉及一种基因缺失突变的荧光检测试剂盒和荧光检测方法,属于化学和生物传感技术领域。
背景技术
基因序列缺失突变是由于dna分子中发生碱基对的缺失引起的基因结构改变,是基因突变中的一种,人体中的基因缺失往往诱发多种疾病,如表皮生长因子(egfr)的18-21号外显子发生基因缺失,诱发肺癌尤其是非小细胞肺癌的产生。伴随着疾病的发生发展,人体病变组织或者血液中缺失突变基因成为临床疾病快速、精确诊断的重要生理指标。目前,qrt-pcr(定量逆转录聚合酶链式反应)和第二代基因测序技术是针对缺失突变基因主要的检测方法。qrt-pcr技术可以实现几个dna分子的扩增,具有很高的灵敏度和专一性,然而主要缺陷在于反应过程需要精确控温,实验条件要求较高,不适合现场使用;第二代基因测序技术需要专业且昂贵的设备进行数据采集和解析,从而限制其广泛应用。
滚环扩增(rollingcircleamplification,rca)是现阶段应用最广泛的恒温扩增技术之一,以环形dna为模板,以寡聚核苷酸(与部分环状模板互补)为引物,在有链置换作用的dna聚合酶(如phi29dnapolymerase)作用下,引物延伸一个循环至起始延伸处置换下旧的dna链而继续进行下一个循环,如此多次循环,生成含有大量重复序列的dna长链。滚环扩增凭借其高特异性、高灵敏度和易操作等性质被大量应用于免疫芯片检测、细胞原位检测、全基因组dna检测、单核苷酸多态性检测等多种领域中。
本发明结合滚环扩增技术、氧化石墨烯荧光淬灭技术和荧光检测法,设计一个针对缺失突变基因的检测体系,其检测限达到1pm,与其他检测缺失突变基因的检测方法相比,具有高灵敏度、高特异性、操作简便、快速的特点。
技术实现要素:
根据本申请的一个方面,提供一种缺失突变基因的荧光检测试剂盒,其特征在于,所述试剂盒包括:pna捕获探针、dna探针、锁式探针、dna聚合酶、滚环扩增引物和荧光探针。
优选地,所述pna捕获探针是与靶基因缺失突变位点附近靠近3’端的10至25个碱基序列完全互补的pna序列,所述pna序列的羧基端与靶基因5’端碱基互补,所述pna序列的氨基端与靶基因3’端碱基互补。
在本发明的一个优选实施方式中,所述的pna捕获探针羧基端(conh2)连接1-5个聚乙二醇单甲醚(mpeg)单体以增加水溶性。
优选地,所述dna探针是与靶基因缺失突变位点两端部分序列完全互补的dna序列;其中,所述dna探针与靶基因杂交时,与靶基因缺失突变位点处对应的部分是以单链形式存在的单链区。
本发明中的dna探针与正常基因杂交形成完全互补的dna双链,而与缺失突变基因杂交时,dna探针中间部分不结合,以单链形式存在。
优选地,所述锁式探针的5’端和3’端与所述dna探针的单链区完全互补,所述锁式探针的5’端和3’端的与所述dna探针单链区的连接点位于所述dna探针单链区的中心。
优选地,所述dna聚合酶和所述滚环扩增引物能够对由所述锁式探针和所述dna探针单链区组成的环状dna进行滚环扩增。
所述锁式探针和所述dna探针单链区在dna连接酶的作用下能够形成环状的dna,以进行后续的滚环扩增。
优选地,所述荧光探针是带有荧光标记的dna信号探针,所述荧光探针能够与所述环状dna的滚环扩增产物上的重复片段序列结合。
优选地,所述dna连接酶选自e·colidna连接酶、t4dna连接酶中的至少一种;更优选为t4dna连接酶。
所述dna聚合酶选自phi29dna聚合酶、t4dna聚合酶、dna聚合酶i中的至少一种;更优选为phi29dna聚合酶。
所述滚环扩增引物选自与环状dna互补配对碱基序列中的至少15个以上碱基。
根据本发明的另一个方面提供使用所述试剂盒进行基因缺失突变荧光检测的方法,所述方法包括以下步骤:
1)将所述pna捕获探针固定在孔板底部,后在缓冲液条件下与靶基因杂交;
2)使被固定在孔板底部的靶基因与dna探针杂交;
3)使与靶基因杂交后的dna探针上的单链部分与锁式探针杂交,并使用连接酶进行环化,在滚环扩增引物和dna聚合酶的作用下进行滚环扩增;
4)使荧光探针与滚环扩增产物杂交,淬灭背景荧光后检测荧光强度的变化。
优选地,所述步骤1)中固定pna捕获探针的方法包括,向孔板加入pna探针和固定液,室温震荡孵育1.5-2.5小时,加入屏蔽液,继续震荡孵育20-40分钟,用缓冲液清洗后加入pbs密封保存备用。
pna捕获探针固定的孔板包括96孔板、48孔板、24孔板、12孔板、6孔板,孔板底部带有活性羧基,在碱性条件下能与氨基反应形成酰胺键。
在本发明的一个优选实施方式中,孵育后分别采用pbst和pbs溶液清洗3次,再加入pbs缓冲液进行保存。
优选地,所述固定液选自碳酸氢钠固定液、碳酸钠固定液、碳酸氢钾固定液、碳酸钾固定液中的至少一种,所述屏蔽液选自赖氨酸屏蔽液、精氨酸屏蔽液、天冬酰胺屏蔽液、谷氨酰胺屏蔽液中的至少一种,所述pna探针的浓度为80-120μm,pna探针和固定液的体积比为1:(80-120)。
优选地,所述步骤1)中,pna捕获探针与靶基因杂交的方法包括,加入阻断液后在室温下震荡孵育20-40分钟,使用缓冲液清洗后加入不同浓度的靶基因,继续震荡孵育后使用缓冲液清洗。
本发明中,使用缓冲液清洗后加入的靶基因,浓度可以包括1fm-100nm。
优选地,所述阻断液选自小牛胸腺dna或鲑鱼精dna的blbs,更优选含有鲑鱼精dna的blbs阻断液。
优选地,所述步骤3)中,环化的方法包括,加入dna连接酶、连接缓冲液和双蒸水在35-40℃的条件下孵育1-2小时后,再于55-65℃的条件下孵育15-25分钟使所述dna连接酶失活。
优选地,所述dna连接酶为t4dna连接酶,所述dna连接酶的浓度为5-9u/μl,所述连接酶缓冲液是10×t4dna连接缓冲液,所述dna连接酶、连接缓冲液和双蒸水的加入体积比为2:(2-4):(24-26)。
优选地,所述步骤3)中滚环扩增的方法包括,加入滚环扩增引物、dna聚合酶、聚合缓冲液、牛血清白蛋白、dntp和双蒸水,在35-40℃下孵育0.5-1.5小时,再于55-65℃的条件下孵育15-25分钟使所述dna聚合酶失活。
优选地,所述dna聚合酶为phi29dna聚合酶,所述聚合酶的浓度为1-5u/μl,所述聚合缓冲液是10×phi29dna反应缓冲液;滚环扩增引物、dna聚合酶、聚合缓冲液、牛血清白蛋白、dntp和双蒸水的加入体积比为1:(1-5):(10-20):1:1:(42-56)。
优选地,所述步骤4)中,荧光探针与所述滚环扩增产物的体积比为1:(20-30),所述荧光探针与所述滚环扩增产物混合孵育1-2小时。
优选地,所述淬灭剂是浓度为80-120μg/ml的氧化石墨烯。
优选地,所述淬灭包括,将荧光探针与滚环扩增产物杂交的产物冷却后,加入淬灭剂、结合缓冲液和双蒸水。
优选地,所述结合缓冲液是10×gobindingbuffer,所述淬灭剂、结合缓冲液和双蒸水的加入体积比为(10-20):10:(50-60)。
本申请能产生的有益效果包括:
1)本申请所提供的试剂盒,能够提供基于滚环扩增技术的基因缺失突变荧光检测中所涉及到的关键试剂。
2)本申请所提供的检测方法,具有高灵敏度、高特异性、操作简便、快速的特点。
附图说明
图1为本发明中检测方法的原理图。
图2为检测9个浓度梯度下egfr缺失突变基因的荧光强度检测结果。
图3为检测9个浓度梯度下egfr缺失突变基因的浓度与荧光强度关系曲线图。
图4为本发明中检测方法的滚环扩增阴性对照实验跑胶结果。
图5为滚环扩增阴性对照实验的荧光强度检测结果。
图6为滚环扩增阴性对照实验中不同对照组的荧光强度对比直方图。
图7为本发明中检测方法的检测探针阴性对照荧光强度检测结果。
图8为检测探针阴性对照实验中不同对照组的荧光强度对比直方图。
图9为在10nmegfr正常基因存在下,不同浓度egfr缺失突变基因的荧光强度检测结果。
图10为在10nmegfr正常基因存在下,不同浓度egfr缺失突变基因的浓度与荧光强度关系曲线图。
具体实施方式
下面结合实施例详述本申请,但本申请并不局限于这些实施例。
如无特别说明,本申请的实施例中的原料均通过商业途径购买。
实施例1
pna捕获探针、dna探针、锁式探针、荧光探针和滚环扩增引物的设计与合成
选择egfr基因缺失位点附近靠近3’端的10-25个碱基序列为对象,设计一条与之完全互补的pna序列作为pna捕获探针,羧基端(conh2)与egfr缺失突变基因5’端碱基互补配对,氨基端(nh2)与egfr缺失突变基因3’端碱基互补配对,羧基端引入1-5个mpeg以增加水溶性,用mbha树脂在hbtu和diea条件下按照碱基序列逐一缩合,经裂解、纯化和结构表征用于后续检测;dna探针、锁式探针和滚环扩增引物由上海杰李生物技术有限公司合成,详细碱基序列如表1所示:
表1egfr缺失突变基因和正常基因、探针以及引物序列
实施例2
egfr缺失突变基因的荧光检测
(1)96孔板上pna捕获探针的固定
每孔中加入1μl100μmpna捕获探针和100μlnahco3固定液,pna捕获探针的终浓度为1.0μm,室温下震荡(600-700rpm)孵育2小时,后加入50μl赖氨酸屏蔽液,继续室温震荡(600-700rpm)0.5小时,用300μl1×pbst清洗四次,250μl1×pbs清洗四次,250μl1×pbs储存备用,用时将250μl1×pbs除去,加入200μlblbs阻断液,室温震荡(600-700rpm)0.5小时,后除去阻断液,直接用于下一步检测;
(2)egfr缺失突变基因的捕获
在100μl体系中加入egfr缺失突变基因和1×pbs缓冲液配置成9个浓度梯度(0fm、1fm、10fm、100fm、1pm、10pm、100pm、1nm、10nm),每个浓度5个重复,室温震荡(600-700rpm)15分钟,将egfr缺失突变基因与pna捕获探针杂交而固定在孔板上,用200μl1×pbs清洗四次;
(3)dna探针与egfr缺失突变基因的杂交
将1μl10μmdna探针加入到99μl1×pbs缓冲液中,与靶基因进行杂交,室温震荡(600-700rpm)15分钟,用200μl1×pbs清洗四次;
(4)锁式探针与dna探针的杂交
将1μl10μm锁式探针加入到99μl1×pbs缓冲液中,与dna探针上未结合的单链区杂交,室温震荡(600-700rpm)15分钟,用200μl1×pbs清洗四次;
(5)t4dna连接酶连接
每组中加入4μl7.0u/μl的t4dna连接酶、10μl10×t4dna连接酶缓冲液和36μl双蒸水,16℃下静置孵育1小时,后置于65℃下20分钟使t4dna连接酶失活;
(6)滚环扩增
每组中加入1μl100μm引物、2μl1.0u/μlphi29dna聚合酶、10μl10×phi29dna聚合反应缓冲液、1μl10mg/mlbsa、1μl10mmdntp、35μl双蒸水,37℃下静置孵育3小时,后置于65℃下20分钟使phi29dna聚合酶失活;
(7)荧光信号检测
取20μl滚环扩增产物与1μl10μm荧光探针在85℃下孵育1分钟,室温震荡(600-700rpm)冷却15分钟,再加入20μl100μg/ml氧化石墨烯、10μl10×氧化石墨烯结合缓冲液和49μl双蒸水,室温震荡(600-700rpm)10分钟,严格避光操作,最后用荧光光谱仪在648nm激发光波长下测量体系发射光谱的变化,并记录不同浓度靶基因存在时的荧光强度。结果如图2和3所示,随着检测egfr缺失突变基因浓度的增加,检测到的荧光强度也随之增强,最低检测灵敏度达到1pm。
实施例3
egfr缺失突变基因荧光检测的阴性对照
(1)96孔板上pna捕获探针的固定
每孔中加入1μl100μmpna捕获探针和99μlnahco3固定液,pna捕获探针的终浓度为1.0μm,室温下震荡(600-700rpm)孵育2小时,后加入50μl赖氨酸屏蔽液,继续室温震荡(600-700rpm)0.5小时,用300μl1×pbst清洗四次,250μl1×pbs清洗四次,250μl1×pbs储存备用,用时将250μl1×pbs除去,加入200μlblbs阻断液,室温震荡(600-700rpm)0.5小时,后除去阻断液,直接用于下一步检测;
(2)egfr基因的捕获
在96孔板中将该实验分成5个组:a、b、c、d、e,其中a、c、d组中加入1μl1μmegfr缺失突变基因,b组中加入1μl1μmegfr正常基因以作阴性对照,e组中不加靶标基因以作空白对照,再分别加入1×pbs缓冲液配置成100μl体系,每组五个重复,室温震荡(600-700rpm)15分钟,将egfr基因与pna捕获探针杂交而固定在孔板上,用200μl1×pbs清洗四次;
(3)dna探针与egfr基因的杂交
在a、b、d和e这四组中分别加入1μl10μmdna探针,c组不加dna探针以作空白对照,每组均加入1×pbs缓冲液配置成100μl体系,与靶基因进行杂交,室温震荡(600-700rpm)15分钟,用200μl1×pbs清洗四次;
(4)锁式探针与dna探针的杂交
在a、b、c和e这四组中分别加入1μl10μm锁式探针,d组不加锁式探针以作空白对照,每组均加入1×pbs缓冲液配置成100μl体系,与dna探针上未结合的单链区杂交,室温震荡(600-700rpm)15分钟,用200μl1×pbs清洗四次;
(5)t4dna连接酶连接
每组中加入4μl7.0u/μl的t4dna连接酶、10μl10×t4dna连接酶缓冲液和36μl双蒸水,16℃下静置孵育1小时,后置于65℃下20分钟使t4dna连接酶失活;
(6)滚环扩增
每组中加入1μl100μm引物、2μl1.0u/μlphi29dna聚合酶、10μl10×phi29dna聚合反应缓冲液、1μl10mg/mlbsa、1μl10mmdntp、35μl双蒸水,37℃下静置孵育3小时,后置于65℃下20分钟使phi29dna聚合酶失活;
(7)聚丙烯酰胺凝胶成像
配制10%的聚丙烯酰胺凝胶(1.5ml40%丙烯酰胺/甲叉双丙烯酰胺、0.6ml10×tae-mg缓冲液、60μl10%aps、8μltemed和3.9ml双蒸水),用凝胶电泳系统在110v电压下跑胶1.5小时,500bpmarker作对照,经gelred染色30分钟,将凝胶置于凝胶成像分析系统中,在紫外灯下检测滚环扩增产物,结果如图4所示,表明egfr缺失突变基因的阳性对照有滚环扩增产物;egfr正常基因作为阴性对照和无靶标基因作为空白对照这两种情况下均无滚环扩增产物;对dna探针和锁式探针也做了空白对照,结果表明均无滚环扩增产物。通过此实验表明该检测体系对egfr缺失突变基因检测的准确性。
(8)荧光信号检测
取20μl滚环扩增产物与1μl10μm荧光探针在85℃下孵育1分钟,室温震荡(600-700rpm)冷却15分钟,再加入20μl100μg/ml氧化石墨烯、10μl10×氧化石墨烯结合缓冲液和49μl双蒸水,室温震荡(600-700rpm)10分钟,严格避光操作,最后用荧光光谱仪在648nm激发光波长下测量体系发射光谱,记录阳性对照、阴性对照和空白对照条件下的各自荧光强度。结果(如图5和6所示)表明阳性对照组的荧光强度最强,平均值达到112.7,而阴性对照组和空白对照组无明显荧光强度,dna探针和锁式探针空白对照组也无明显荧光强度,该荧光信号检测结果与跑胶结果相吻合,再次证明该检测体系应用于egfr缺失突变基因检测的准确性和实用性。
实施例4
egfr缺失突变基因荧光检测的检测探针阴性对照
该实验分成5个组:1、2、3、4、5,每组中均加入1μl10μm荧光探针,1和2组中不加任何检测基因,3、4和5组中分别加入20μl10nmegfr缺失突变基因滚环扩增产物、20μl10nmegfr正常基因滚环扩增产物和1μl10μm锁式探针,每组5个重复,在85℃下孵育1分钟,室温震荡(600-700rpm)冷却15分钟,每组加入10μl10×氧化石墨烯结合缓冲液,除1组外其他四组均加入20μl100μg/ml氧化石墨烯,最后加入双蒸水配置成100μl检测体系。室温震荡(600-700rpm)10分钟,严格避光操作,用荧光光谱仪在648nm激发光波长下测量体系发射光谱,记录阳性对照、阴性对照和空白对照条件下的各自荧光强度。结果如图7和8所示,这表明在无检测基因和氧化石墨烯存在的情况下100nm荧光探针的荧光强度平均值为295.22,阳性对照组的荧光强度平均值为112.7,而阴性对照组(4和5组)以及空白对照组(2组)无明显荧光强度,这不仅说明荧光探针在100nm浓度下足够检测滚环扩增产物,而且也证明该荧光探针可用于egfr缺失突变基因滚环产物的荧光检测。
实施例5
混合基因中egfr缺失突变基因的荧光检测
(1)96孔板上pna捕获探针的固定
每孔中加入1μl100μmpna捕获探针和100μlnahco3固定液,pna捕获探针的终浓度为1.0μm,室温下震荡(600-700rpm)孵育2小时,后加入50μl赖氨酸屏蔽液,继续室温震荡(600-700rpm)0.5小时,用300μl1×pbst清洗四次,250μl1×pbs清洗四次,250μl1×pbs储存备用,用时将250μl1×pbs除去,加入200μlblbs阻断液,室温震荡(600-700rpm)0.5小时,后除去阻断液,直接用于下一步检测;
(2)egfr基因的捕获
9个浓度梯度的egfr缺失突变基因均混合1μl1μmegfr正常基因,用1×pbs缓冲液配置成0fm、1fm、10fm、100fm、1pm、10pm、100pm、1nm、10nm的100μl体系,每个浓度5个重复,室温震荡(600-700rpm)15分钟,将egfr混合基因与pna捕获探针杂交而固定在孔板上,用200μl1×pbs清洗四次;
(3)dna探针与egfr混合基因的杂交
将1μl10μmdna探针加入到99μl1×pbs缓冲液中,与egfr混合基因进行杂交,室温震荡(600-700rpm)15分钟,用200μl1×pbs清洗四次;
(4)锁式探针与dna探针的杂交
将1μl10μm锁式探针加入到99μl1×pbs缓冲液中,与dna探针上未结合的单链区杂交,室温震荡(600-700rpm)15分钟,用200μl1×pbs清洗四次;
(5)t4dna连接酶连接
每组中加入4μl7.0u/μl的t4dna连接酶、10μl10×t4dna连接酶缓冲液和36μl双蒸水,16℃下静置孵育1小时,后置于65℃下20分钟使t4dna连接酶失活;
(6)滚环扩增
每组中加入1μl100μm引物、2μl1.0u/μlphi29dna聚合酶、10μl10×phi29dna聚合反应缓冲液、1μl10mg/mlbsa、1μl10mmdntp、35μl双蒸水,37℃下静置孵育3小时,后置于65℃下20分钟使phi29dna聚合酶失活;
(7)荧光信号检测
取20μl滚环扩增产物与1μl10μm荧光探针在85℃下孵育1分钟,室温震荡(600-700rpm)冷却15分钟,再加入20μl100μg/ml氧化石墨烯、10μl10×氧化石墨烯结合缓冲液和49μl双蒸水,室温震荡(600-700rpm)10分钟,严格避光操作,最后用荧光光谱仪在648nm激发光波长下测量体系发射光谱的变化,并记录不同浓度靶基因存在时的荧光强度。结果如图9和10所示,随着egfr缺失突变基因浓度的增加,检测到的滚环扩增产物荧光强度也随之增强,最低检测灵敏度达到1pm。这表明在10nmegfr正常基因存在下该检测方法依然准确、有效和可靠。该实验的设计模拟了真实的检测样本情况,为患者非小细胞肺癌组织中egfr缺失突变基因的检测奠定基础。
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
序列表
<110>中国科学院宁波工业技术研究院慈溪生物医学工程研究所
中国科学院宁波材料技术与工程研究所
<120>基因缺失突变的荧光检测试剂盒和荧光检测方法
<130>20180627
<141>2018-06-27
<160>5
<170>siposequencelisting1.0
<210>1
<211>50
<212>dna
<213>artificiallysynthesized
<400>1
ttcccgtcgctatcaacgaaagccaacaaggaaatcctcgatgtgagttt50
<210>2
<211>55
<212>dna
<213>artificiallysynthesized
<400>2
tccttgttggctttcggagatgttgcttctcttaattccttgatagcgacgggaa55
<210>3
<211>98
<212>dna
<213>artificiallysynthesized
<400>3
aagcaacatctcaactatcataagactcgtcatgtctcagcagcttctaacggtcactaa60
tacgactcactataggttctggaaagcggaattaagag98
<210>4
<211>18
<212>dna
<213>artificiallysynthesized
<400>4
gctgagacatgacgagtc18
<210>5
<211>18
<212>dna
<213>artificiallysynthesized
<400>5
ctaacggtcactaatacg18
- 还没有人留言评论。精彩留言会获得点赞!