聚乙二醇修饰药物运输荧光高分子材料及其制备方法与流程
本发明属于超分子自组装领域,具体涉及一种聚乙二醇修饰的双溴取代氟硼吡咯材料及其制备方法。
背景技术
近年来伴随着生物医学的快速发展,对于抗癌新方法的探究成为了科学关注的焦点之一。由于现代社会癌症的病发率日益增长,对于癌症的预防,治疗手段也日益趋于多样化。目前对于癌症的治疗方法还是以传统的化疗手段为主,其他新型治疗方式也包括放疗,手术切除,基因疗法,光动力治疗等。化疗有其方便性和有效性,但是化疗对于患者造成的副作用大这一现象同样也成为了困扰临床医学的难题,而基因疗法受限于当代的医疗条件未能广泛的普及应用。
光动力治疗法作为一种新型的抗癌治疗手段,有创伤小,适用于局部治疗等多种特点,使得该方法受到了更多的关注。由于人体内水分含量很高,而现阶段的传统光动力治疗方法中大多数光敏剂药物都具有疏水性的特点,因此,要构建一种安全的具有亲水性特点且可以携带多种不同药理作用的药物载体,能够实现多种治疗方法,这样势必可以提高癌症的治愈率。
技术实现要素:
本发明的目的是提供一种聚乙二醇修饰药物运输的荧光高分子及其制备方法。
实现本发明目的的技术解决方案是:
一种聚乙二醇修饰药物运输的荧光高分子,其结构式(m3)如下所示:
式中,n=45。
本发明还提供了所述产物的中间体m1和m2,其结构式分别如下:
上述中间产物m1的制备方法,包括如下步骤:在无水乙醇体系下,将化合物a和水合肼发生回流反应制备中间体m1的步骤;
优选的,水合肼为质量85%的水合肼,水合肼和化合物a的摩尔比为6:1-8:1,反应回流为至少两个小时。
上述中间产物m2的制备方法,包括如下步骤:以乙腈为溶剂,以无水碳酸钾为碱性试剂,将mpeg-ots和对羟基苯甲醛在氩气保护下发生回流反应制备中间体m2的步骤;
优选的,碳酸钾和对羟基苯甲醛的摩尔比为7:1,对羟基苯甲醛和mpeg-ots的摩尔比为10:7,回流反应时间为三天。
目标产物m3的制备方法,包括如下步骤:在甲醇体系下,将中间体m1和中间体m2在80±5℃下发生反应制备目标化合物m3的步骤;
优选的,m1和m2的摩尔比为1.1:1,反应时间至少在8个小时以上。
上述目标产物m3作为自组装超分子材料,及其制备光动力治疗和抗癌药物载体的上应用。
本发明与现有技术相比,其优点有:
(1)聚乙二醇修饰药物运输的荧光高分子产率较高,合成简单。(2)聚乙二醇修饰药物运输的荧光高分子具有良好的水溶性,适用于人体水环境。
附图说明
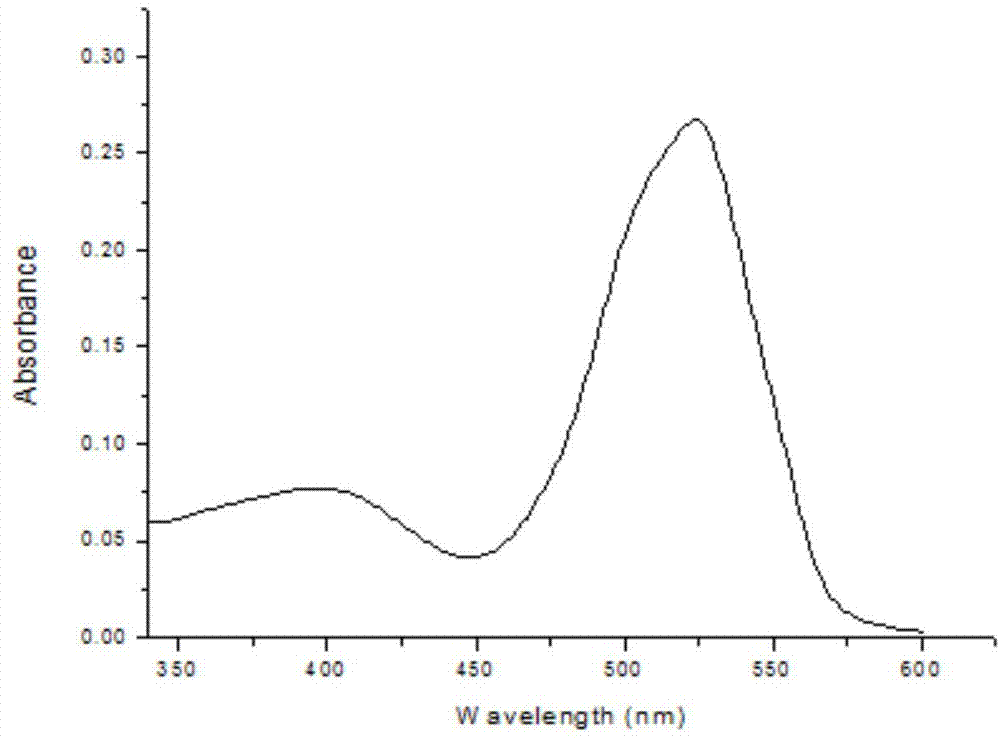
图1为m3的紫外吸收光谱测定曲线。
图2为孟加拉玫瑰红的紫外吸收光谱测定曲线。
图3为m3单线态氧的变化曲线。
图4为孟加拉玫瑰红单线态氧的变化曲线。
图5为m3分子在酸性环境下产生单线态氧的变化曲线。
图6为m3分子行阿霉素药物运输在不同ph下的释放。
具体实施方式
(一)聚乙二醇修饰药物运输的荧光高分子的合成路线如下:
上述聚乙二醇修饰药物运输的荧光高分子(目标产物m3)的制备方法,包括以下步骤:
步骤一:将对羟基苯甲醛和无水碳酸钾溶于丙酮并置于烧瓶中,搅拌使碳酸钾固体在溶液中分散均匀。氯乙酸乙酯恒压滴加,将反应器置于油浴锅内,搅拌回流8小时后得到化合物1;
步骤二:将化合物1和2,4-二甲基吡咯置于容器中,加入溶剂二氯甲烷后,滴加催化剂三氟乙酸,在室温下搅拌过夜,再加入四氯苯醌,继续搅拌5小时以上,滴加三乙胺和三氟化硼乙醚后继续反应得到化合物2;
步骤三:将化合物2和四氯化碳置于反应器中,并将反应器置于油浴锅内,滴加一滴dmf做催化剂,待c1完全溶解后,将n-溴代琥珀酰亚胺分批加入反应体系中。将反应温度升高至85℃回流反应,保持回流反应2小得到红色固体物质3;
步骤四:在三口烧瓶中加入化合物3,再向三口烧瓶中加入无水乙醇,将反应容器置于油浴锅内,调整油浴锅温度,待c2完全溶解后,向三口烧瓶中逐滴加入过量的浓度为85%的水合肼回流反应以得到橙红色固体物质m1;
步骤五:在三口烧瓶中加聚乙二醇单甲醚(mpeg-2000),再向三烧瓶中加入四氢呋喃,将反应容器放置于冰水浴中。氢氧化钾固体溶于去离子水中,在冰水浴条件下用恒压滴液漏斗逐滴滴加溶液至四氢呋喃溶液中,体系保持冰水浴条件继续反应。称取对甲苯磺酰氯溶于四氢呋喃中,在冰水浴条件下用恒压滴液漏斗逐滴滴加至反应四氢呋喃/水的体系当中,保持边滴加边搅拌状态,约2小时后滴加完毕。加料完毕后保持冰水浴反应4小时得到化合物mpeg-ots;
步骤六:在洁净的三口烧瓶中加入mpeg-ots,再向三口烧瓶中加入乙腈,搅拌溶解。将无水碳酸钾固体加入到乙腈溶液中,继续搅拌10分钟。称取对羟基苯甲醛加入到反应体系中,在氩气保护下回流反应得到化合物m2;
步骤七:三口烧瓶中加入m1,加入甲醇溶解,称取m2加入到三口烧瓶中搅拌溶解。将三口烧瓶置于油浴锅中,将油浴锅温度升至80℃,使混合物回流反应。保持回流状态过夜反应,得到化合物m3。
实施例1:聚乙二醇修饰药物运输的荧光高分子的合成
1.化合物[1]的合成
在洁净的250ml三口烧瓶中,加入对羟基苯甲醛(12.2g,100mmol),再向三口烧瓶中加入150ml经干燥处理后的丙酮。待对羟基苯甲醛完全溶解之后,向反应容器中加入无水碳酸钾固体(20.7g,150mmol),搅拌使碳酸钾固体在溶液中分散均匀。称取18.3g氯乙酸乙酯于50ml的恒压滴液漏斗中,在室温条件下(25℃)向三口烧瓶内逐滴滴加氯乙酸乙酯,约1小时后滴加完成,撤去恒压滴液漏斗,将反应器置于油浴锅内,在油浴温度65摄氏度条件下搅拌回流反应8个小时后冷却至室温。混合物过滤,得无色透明的溶液,在旋转蒸发仪上去除溶剂,得到剩余固体。将固体残留物溶于100ml的乙酸乙酯当中。配置1mol/l的氢氧化钾溶液200ml,在分液漏斗中用氢氧化钾溶液洗涤乙酸乙酯层两次(2×100mll,留取上层有机层再用去离子水水洗洗一次,最后用饱和的氯化钠溶液洗涤一次,收集有机层,加入无水硫酸镁静置干燥2小时。过滤,滤饼用乙酸乙酯多次洗涤得到产物的乙酸乙酯溶液。用旋转蒸发仪在水浴温度为55℃的条件下将溶剂除去,残留物放置于鼓风干燥烘箱内设定温度为50摄氏度进行鼓风干燥2小时。得到无色透明的油状产物1(16.2g,77.9mmol),产率为77.9%。
2.化合物[2]的合成
取一洁净的500ml三口烧瓶,多次抽真空,在氩气保护下向三口烧瓶中加入化合物1(4.5g,21.64mmol)和2,4-二甲基吡咯(3.8g,40mmol),再在三口烧瓶中加入300ml重蒸过的二氯甲烷,在室温下搅拌溶解。滴入几滴三氟乙酸作为催化剂使原料进行缩合反应,当溶液由无色慢慢变为紫红色时,用锡箔纸将反应容器包裹住进行避光处理,并多次通入氩气,保持氩气氛围,反应进行12个小时。通过tcl法跟踪反应进程,当原料a1已经不再减少时,进行下一步操作。
上述反应在氩气保护环境下,将3.9g四氯苯醌溶于50ml重蒸过的二氯甲烷中,用恒压滴液漏斗缓慢滴入反应器中,持续搅拌,避光处理,并持续通氩气。滴加完成后继续反应4小时。再用量筒量取20ml的三乙胺置于恒压滴液漏斗中,逐滴滴加至三口烧瓶中,滴加完毕后,用恒压滴液漏斗将20ml三氟化硼乙醚逐滴滴加至反应反应体系当中。滴加完毕后,反应体系在室温(25℃)条件下继续搅拌反应12个小时,停止反应。
上述反应体系用去离子水多次水洗,萃取所得的水层再用二氯甲烷萃取三次,合并有机层,收集的有机层用无水硫酸镁干燥,静置两小时。过滤,滤饼用二氯甲烷多次冲洗,所得溶液在旋转蒸发仪上除去溶剂,拌硅胶。湿法上样,用展开剂(pe:dcm=10:1~pe:dcm=4:1)对其进行柱层析分离,得到深红色固体物质(2.5g,5.87mmol),产率为39.35%。
红色固体,产率39.35%,熔点210~212℃。
1h-nmr(500mhz,cdcl3)δ(tms,ppm):7.16(d,j=8.1hz,2h),7.01(d,j=8.1hz,2h),5.96(s,2h),4.67(s,2h),4.28(q,j=7.1hz,2h),2.53(s,6h),1.40(s,6h),1.33-1.18(m,3h).
13c-nmr(126mhz,cdcl3)δ(tms,ppm):167.44,157.30,154.28,142.00,140.29,130.63,128.22,127.19,127.02,126.74,120.09,114.22,64.32,60.40,13.46,13.43,13.06.
esi-ms:m/z=426.99[m+h+].
3.化合物[3]的合成
在洁净的100ml三口烧瓶中加入化合物[2](0.43g,1mmol),再向三口烧瓶中加入20ml处理过的四氯化碳,搅拌。将反应器置于油浴锅内,调整油浴锅温度,将温度升至50℃,继续搅拌溶解,滴加一滴dmf做催化剂,待c1完全溶解后,称取n-溴代琥珀酰亚胺(0.43g,2.4mmol)分批加入反应体系中。将反应温度升高至85℃回流反应,保持回流反应2小时。关闭加热装置,停止反应。过滤,滤液在旋转蒸发仪上除去溶剂,拌硅胶。湿法上样,用展开剂(pe:dcm=8:1~5:1)对其进行柱层析分离,得到红色固体物质(0.4g,0.69mmol),产率为68.7%。
红色固体,产率68.7%,熔点271~273℃。
1h-nmr(500mhz,cdcl3)δ(tms,ppm):7.21-7.17(m,2h),7.08(d,j=8.7hz,2h),4.73(s,2h),4.32(d,j=7.1hz,2h),2.62(s,6h),1.43(s,6h),1.33(t,j=7.1hz,3h).
4.化合物m1的合成
在洁净的100ml三口烧瓶中加入化合物[3](0.4g,0.69mmol),再向三口烧瓶中加入20ml无水乙醇,搅拌。将反应容器置于油浴锅内,调整油浴锅温度,将温度升至50℃,继续搅拌溶解,待化合物[3]完全溶解后,向三口烧瓶中逐滴加入过量的浓度为85%的水合肼。将反应温度升至90℃进行回流反应,保持回流反应2小时。用tlc法跟踪反应进程,当原料完全消失后停止反应。将反应液冷却至室温,混合层用二氯甲烷萃取多次,有机层再用等体积的去离子水水洗两次,最后再用饱和食盐水洗涤一次。收集有机层,用无水硫酸钠干燥两小时,过滤,滤饼用二氯甲烷多次冲洗,得到橙红色透明澄清溶液。用旋转蒸发仪将溶液中的溶剂除去,自然晾干。得到橙红色固体物质m1(0.31g,0.55mmol),产率为80%。
红色固体,熔点50~51℃。
1h-nmr(500mhz,cdcl3)δ(tms,ppm):8.23(s,0.5h),7.85(s,0.5h),7.69(d,j=8.4hz,1h),7.61(d,j=8.4hz,1h),7.16(qd,j=7.4,6.8,3.5hz,3h),7.01–6.89(m,3h),5.22(s,1h),4.75(d,j=2.6hz,1h),4.18(dt,j=5.0,3.0hz,2h),3.88(q,j=4.2hz,2h),3.64(d,j=3.4hz,202h),3.38(s,3h),2.61(d,j=2.9hz,6h),1.43(d,j=11.5hz,6h).
5.化合物mpeg-ots的合成
在三口烧瓶中加入20g(约10mmol)聚乙二醇单甲醚(mpeg-2000),再向三口烧瓶中加入50ml四氢呋喃,待聚乙二醇单甲醚完全溶解后,将反应容器放置于冰水浴(0℃)中,继续搅拌。称取20g氢氧化钾固体溶于50ml去离子水中,在冰水浴条件下用恒压滴液漏斗逐滴滴加溶液至四氢呋喃溶液中,约一个小时滴加完毕,体系保持冰水浴条件继续反应。称取对甲苯磺酰氯(3.8g,20mmol)溶于50ml四氢呋喃中,溶液放置于恒压滴液漏斗之中,在冰水浴条件下用恒压滴液漏斗逐滴滴加至反应四氢呋喃/水的体系当中,保持边滴加边搅拌状态,约2小时后滴加完毕。加料完毕后保持冰水浴反应4小时。撤去冰水浴,反应体系在室温下继续反应8小时。向反应液中加入冰水淬灭反应,混合液用二氯甲烷多次萃取,合并有机层,有机层用去离子水水洗两次,再用饱和食盐水洗涤。收集有机层,用无水硫酸钠干燥。过滤,滤饼用二氯甲烷冲洗,得到无色澄清溶液。用旋转蒸发仪上除去溶剂,得到无色油状液体,将无色油状液体加入到过量的无水乙醚当中,有大量的白色固体沉淀析出,减压过滤,滤饼用无水乙醚多次洗涤,将滤饼自然晾干,得到白色固体产物mpeg-ots(15.6g,约7.4mmol),产率约为74.2%。
白色固体,产率74.2%,熔点35~37℃。
1h-nmr(500mhz,cdcl3)δ(tms,ppm):7.69(dd,j=8.3,3.4hz,2h),7.30-7.20(m,2h),4.05(s,2h),3.71-3.65(m,2h),3.54(d,j=4.1hz,180h),3.27(d,j=3.6hz,3h),2.35(d,j=3.0hz,3h).
6.化合物[m2]的合成
在三口烧瓶中加入mpeg-ots(15g,约7.1mmol),再向三口烧瓶中加入300ml乙腈,搅拌溶解。称取无水碳酸钾固体(10g,72.5mmol)加入到乙腈溶液中,继续搅拌10分钟。称取对羟基苯甲醛(1.3g,10.65mmol)加入到反应体系中,搅拌溶解。将三口烧瓶置于油浴锅中,将温度升高至90℃,在氩气保护下回流反应3天。停止反应,过滤,滤饼用乙腈冲洗,得到的溶液用旋转蒸发仪除去溶剂。残留物用二氯甲烷萃取两次,收集有机层并用饱和食盐水洗涤一次。得到的有机层用无水硫酸镁干燥。过滤,滤液用旋转蒸发仪除去大部分溶剂,浓缩后的溶液倒入过量的无水乙醚当中,搅拌数分钟,有白色固体产生。减压过滤,滤饼用无水乙醚多次冲洗,滤饼自然晾干,得到白色固体物质m2(11.6g,约5.5mmol)。产率约为77.3%。
白色固体,产率56%,熔点42~44℃。
1h-nmr(500mhz,dmso-d6)δ(tms,ppm):9.80(s,1h),7.75(d,j=8.5hz,2h),6.95(d,j=8.5hz,2h),4.14(t,j=4.8hz,2h),3.81(t,j=4.8hz,2h),3.56(s,201h),3.30(s,3h).
7.目标产物[m3]的合成
在100ml三口烧瓶中加入m1(0.3g,0.55mmol),再加入20ml甲醇溶解,称取m2(1.1g,约0.52mmol)加入到三口烧瓶中搅拌溶解。将三口烧瓶置于油浴锅中,将油浴锅温度升至80℃,使混合物回流反应。过夜反应后停止加热,趁热将反应体系放置于冰水浴中,有固体析出,过滤,用无水乙醚洗涤固体三次,再用石油醚多次洗涤固体,得到红色固体m3。
红色固体,产率61%,熔点50~51℃。
1h-nmr(500mhz,cdcl3)δ(tms,ppm):8.23(s,0.5h),7.85(s,0.5h),7.69(d,j=8.4hz,1h),7.61(d,j=8.4hz,1h),7.16(qd,j=7.4,6.8,3.5hz,3h),7.01–6.89(m,3h),5.22(s,1h),4.75(d,j=2.6hz,1h),4.18(dt,j=5.0,3.0hz,2h),3.88(q,j=4.2hz,2h),3.64(d,j=3.4hz,202h),3.38(s,3h),2.61(d,j=2.9hz,6h),1.43(d,j=11.5hz,6h).
目标产物m3在水中溶解度较好,并且溶于大多数有机溶剂。
实施例2:紫外吸收光谱测定
(1)目标产物m3分子的紫外吸收光谱测定
用移液枪吸取2mlm3分子的待测液(0.01mmol/l)转移至4ml的比色皿中。预设紫外吸收波长范围确定在300nm至600nm之间,先做空白试验以此来扣除所用溶剂的空白干扰,最后再进行紫外吸收光谱测定。
(2)孟加拉玫瑰红的紫外吸收光谱测定
用移液枪吸取2ml孟加拉玫瑰红分子的待测液(0.01mmol/l)转移至4ml的比色皿中。预设紫外吸收波长范围确定在300nm至600nm之间,先做空白试验以此来扣除所用溶剂的空白干扰,最后再进行紫外吸收光谱测定。
对比图1和图2吸光度,我们可以发现,m3分子的紫外最大吸光波长在523nm左右,而孟加拉玫瑰红的紫外最大吸光波长在550nm左右,两者的最大吸光波长接近,且两者的吸光度数值也比较大,所以选择孟加拉玫瑰红作为单线态产率测试的参比样,符合我们参比样品的实验要求。
实施例3:单线态氧变化测定
(1)目标产物m3分子的单线态氧变化测定
用移液枪吸取2mlm3分子的待测液(0.01mmol/l)转移至4ml的比色皿中。测定其吸收光谱后,用移液枪逐步移取一定量的配置好的1,3-二苯基异苯并呋喃(dpbf)溶液(10mmol/l)并滴加到装有m3分子的待测液(0.01mmol/l)的比色皿中,并测试其吸光度,当在410nm处的吸光度达到0.5以上时停止滴加dpbf溶液。对混合物进行单线态氧的测定。通过一定功率和一定波长的激光照射比色皿中的溶液,多次照射,照射时间相等且每次照射的时间间隔一致,分别测试每次照射完成后待测混合液的紫外吸收光谱。
(2)孟加拉玫瑰红分子的单线态氧变化测定
用移液枪吸取2ml孟加拉玫瑰红分子的待测液(0.01mmol/l)转移至4ml的比色皿中。测定其吸收光谱后,用移液枪逐步移取一定量的配置好的1,3-二苯基异苯并呋喃(dpbf)溶液(10mmol/l)并滴加到装有孟加拉玫瑰红分子的待测液(0.01mmol/l)的比色皿中,并测试其吸光度,当在410nm处的吸光度达到0.5以上时停止滴加dpbf溶液。对混合物进行单线态氧的测定。通过一定功率和一定波长的激光照射比色皿中的溶液,多次照射,照射时间相等且每次照射的时间间隔一致,分别测试每次照射完成后待测混合液的紫外吸收光谱。
对比图3和图4的吸光度变化曲线,我们可以发现,m3分子的混合溶液在相同的照射条件下,在410nm处的吸光度数值下降速度要明显大于孟加拉玫瑰红的混合溶液,这一现象也定性说明我们制备的m3分子不仅具有良好的水溶性,而且产生单线态氧的效率也十分的可观,测试性能要优于孟加拉玫瑰红。
实施例4:m3分子在酸性环境下产生单线态氧的变化曲线
通过观察附图5的变化曲线我们发现,m3分子在酸性环境下在410nm处的吸光度数值下降速度要明显快于其在中性去离子水中的下降速度,在光照射4次共20s后,溶液在410nm处的紫外吸光度就趋于平缓,达到临界值附近。证明在激光照射,弱酸性的环境对m3分子产生单线态氧具有促进作用,释放速率和产生效率均要高于中性环境。证明我们制备的光敏剂药物并不会因为酸性条件而失活反而在酸性环境下有促进作用,具有应用到人体内药物测试的开发前景。
实施例5:m3进行阿霉素药物运输在不同ph下的释放率
通过分析图6,我们发现酸性条件下比中性条件下荧光强度增强,说明原本包裹着阿霉素分子的m3分子已经释放出了阿霉素,两者之间的相互作用键断裂,使得部分m3恢复了自身荧光,导致荧光增强,而且随着时间的推移,荧光强度逐步增强,说明在逐步释放出阿霉素,而且我们发现600nm处阿霉素发射波长的荧光强度也在逐渐增大,说明阿霉素的含量在逐步升高,有阿霉素释放产生,证明m3是良好的药物载体适用于人体环境,可以进行药物运输。
- 还没有人留言评论。精彩留言会获得点赞!