含磷丙烯酸单体和阻燃型相变微胶囊及其制备方法与流程
本发明属于节能材料
技术领域:
,涉及含磷丙烯酸单体、阻燃型相变微胶囊及其制备方法。
背景技术:
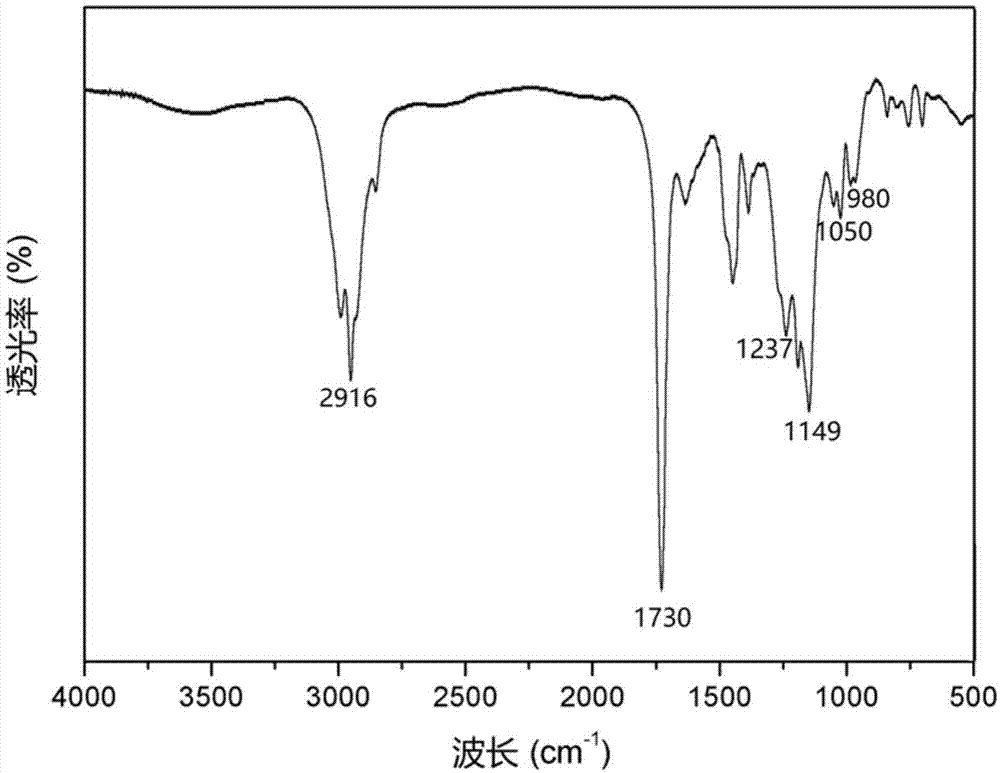
近年来,伴随着人类社会能源消耗的与日俱增及地球环境问题的日益恶化,基于相变储能材料(pcm)的热能存储系统(tes)引起了全球科学工作者的广泛关注。相变储能材料可以在特定的温度范围内利用相变过程来吸收、储存、释放大量的热量,从而实现蓄热调温作用,被广泛应用于节能建筑、智能纺织品、电力调峰、太阳能存储以及工业废热回收等领域。有机相变材料储能密度高、相变温度适宜、耐热稳定性好,具有无机相变材料无法比拟的优点,是目前相变储能领域研究的重点。然而,有机相变材料通常具有较差的导热性能,并且其相变过程为固-液相变,在热能存储过程中易发生泄漏,限制了其在实际应用中的进一步推广。微胶囊化相变材料可以克服有机相变材料的诸多局限,通过表面聚合物包覆,防止了相变材料的泄漏,并且微胶囊具有极高的比表面积,增大了热能存储和释放效率,极大地拓宽了相变材料的应用范围。高分子聚合物具有良好的物理机械性能,有利于承受相变材料在使用过程中体积的反复变化,因此微胶囊相变材料通常以脲醛树脂、三聚氰胺甲醛树脂、丙烯酸树脂、聚氨酯聚脲树脂等有机聚合物作为壁材。然而,由于有机相变材料及传统聚合物壁材均为易燃物质,限制了相变微胶囊在建筑及纺织品等领域的应用。近年来,火灾频繁发生对人类的生命和财产安全造成了极大的危害,建筑及纺织品等领域对材料的防火等级提出了新的要求,因此阻燃型相变储能材料成为了目前相变储能领域的研究重点。中国专利cn105542721a公开了一种阻燃相变微胶囊的制备方法,通过向有机相变材料中混入阻燃剂氯化石蜡制备了阻燃相变微胶囊,但是阻燃剂的加入降低了微胶囊中相变材料的含量,从而降低了相变微胶囊的储能密度。中国专利cn105112020a公开了另外一种阻燃相变微胶囊的制备方法,通过将反应型膦酸酯单体与季戊四醇四丙烯酸酯反应得到具有一定阻燃性能的聚合物囊壁,但是膦酸酯单体与季戊四醇四丙烯酸酯的反应过程难以控制,操作难度大。中国专利cn103740337a将甲基丙烯酸依次与丁二醇和五氧化二磷反应制备了一种膦酸酯功能性单体,并通过其与丙烯酸单体聚合制备了另外一种阻燃微胶囊材料,但是该膦酸酯功能性单体制备过程繁琐,容易产生副产物,并且所制备的相变微胶囊囊壁交联密度低,易导致微胶囊的使用稳定性下降。技术实现要素:本发明目的在于克服现有技术的不足,提供含磷丙烯酸单体和囊壁为含磷丙烯酸单体改性丙烯酸树脂的阻燃型相变微胶囊及其制备方法,以便在保证阻燃性能的前提下,提高相变微胶囊的储能密度和囊壁的交联密度并简化制备工艺。本发明所述含磷丙烯酸单体,由n,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯与含双键酰氯通过michael加成反应制得。所述含双键酰氯为丙烯酰氯、甲基丙烯酰氯或2-丁烯酰氯,当含双键酰氯为丙烯酰氯时,含磷丙烯酸单体的结构式如下:当含双键酰氯为甲基丙烯酰氯时,含磷丙烯酸单体的结构式如下:当含双键酰氯为2-丁烯酰氯时,含磷丙烯酸单体的结构式如下:本发明所述阻燃型相变微胶囊,由囊壁和包覆于囊壁中的有机相变物质构成,所述囊壁为含磷丙烯酸单体改性丙烯酸树脂,所述含磷丙烯酸单体改性丙烯酸树脂通过上述含磷丙烯酸单体与丙烯酸单体反应形成。本发明所述阻燃型相变微胶囊,有机相变物质为通式是cnh2n+2的直链烷烃或支链烷烃、通式是cnh2ncooh的高级脂肪酸、通式是cnh2noh的高级脂肪醇中的至少一种,上述通式中,n=12~32。本发明所述阻燃型相变微胶囊其形状为球形或类球形,平均粒径为120~150nm。本发明所述含磷丙烯酸单体的制备方法,步骤如下:将n,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于溶剂a后,加入中和剂及含双键酰氯并混合均匀形成反应体系,然后在10℃~50℃搅拌反应4~24h,反应时间到达后,将反应液过滤得滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的含双键酰氯和中和剂,再通过减压蒸馏除去萃取所得油相中的溶剂a,即得到含磷丙烯酸单体;所述反应体系中,n,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯、含双键酰氯与中和剂的摩尔比为1:(1~3):(1~3),溶剂a的质量与n,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯、中和剂和含双键酰氯总质量之比为(5~20):1。上述含磷丙烯酸单体的制备方法中,所述中和剂为三乙胺、质量浓度25~28%的氨水、氢氧化钠或氢氧化钾;所述含双键酰氯为丙烯酰氯、甲基丙烯酰氯或2-丁烯酰氯;所述溶剂a为二氯甲烷、二氯乙烷、三氯甲烷、三氯乙烷、四氯甲烷或四氯乙烷。上述方法中,萃取时饱和氯化钠溶液与滤液得质量比为(1~3):1。本发明所述阻燃型相变微胶囊的制备方法,步骤如下:(1)在有机相变物质熔点温度或不高于有机相变物质熔点温度20℃的条件下,将上述方法制备的含磷丙烯酸单体与丙烯酸单体、引发剂、有机相变物质搅拌混合均匀得到油相,在室温下将去离子水和乳化剂搅拌混合均匀得到水相;所述含磷丙烯酸单体与丙烯酸单体的质量比为(0.05~0.5):1,引发剂、有机相变物质与含磷丙烯酸单体和丙烯酸单体总质量之比为(0.001~0.03):(0.3~2):1,去离子水、乳化剂与油相的质量比为(3~8):(0.005~0.05):1。(2)在搅拌下于有机相变物质熔点温度或不高于有机相变物质熔点温度20℃,将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在1000~3000r/min的转速条件下搅拌10~60min,再于60~90℃以200~800r/min的转速搅拌反应1~8h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼,然后将洗涤后的滤饼真空干燥得到阻燃型相变微胶囊。上述阻燃型相变微胶囊的制备方法中,所述丙烯酸单体为丙烯酸、甲基丙烯酸、苯乙烯、甲基苯乙烯、丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丁酯、丙烯酸异丁酯、甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸异丁酯、甲基丙烯酸正丁酯中的至少一种;所述有机相变物质为通式是cnh2n+2的直链烷烃或支链烷烃、通式是cnh2ncooh的高级脂肪酸、通式是cnh2noh的高级脂肪醇中的至少一种,上述通式中,n=12~32。上述阻燃型相变微胶囊的制备方法中,所述引发剂为偶氮二异丁腈、偶氮二异庚腈或过氧化二苯甲酰;所述乳化剂为十二烷基磺酸钠、十二烷基苯磺酸钠、十二烷基硫酸钠、tween-20、tween-40、tween-60、tween-80、span-20、span-40、span-60、span-80、tritonx-100中的至少一种。本发明具有如下有益效果:1、本发明利用n,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯与含双键酰氯通过michael加成反应,制备了含两个碳碳双键的含磷丙烯酸单体,再以含磷丙烯酸单体为交联剂与丙烯酸单体进行聚合反应,通过包裹有机相变材料,制备了囊壁具有阻燃性能的相变微胶囊,由于相变微胶囊的囊壁为含磷丙烯酸单体改性丙烯酸树脂,因而本发明提供的相变微胶囊不仅具有优良的阻燃性能,而且囊壁的交联密度提高,相变焓的损失率更小,使用稳定性更好,2、由于本发明所述阻燃型相变微胶囊不含阻燃剂,因而确保了相变微胶囊具有较高的储能密度和储能能力。3、本发明所述含磷丙烯酸单体由n,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯与含双键酰氯通过michael加成反应制得,阻燃型相变微胶囊由自由基乳液聚合法制得,因而反应过程易控,制备工艺简便,生产周期短,易于工业化生产。附图说明图1为实施例1所制备阻燃型相变微胶囊的红外谱图,谱图中,2916cm-1处为c-h的伸缩振动峰,1730cm-1和1149cm-1处为酯键的伸缩振动峰,1237cm-1和1050-980cm-1处的吸收峰分别是由p=o和p-o-c基团产生的。图2为实施例1、实施例2所制备阻燃型相变微胶囊的dsc曲线图。图3为实施例1、实施例2所制备阻燃型相变微胶囊的sem照片(放大倍数5万倍)。具体实施方式下面通过实施例对本发明所述含磷丙烯酸单体和相变微胶囊及其制备方法作进一步说明。在此有必要指出的是以下实施例只是用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制。下述实施例中,红外光谱分析采用美国perkin–elmer公司ftir170型傅立叶变换红外光谱仪测定样品的红外光谱;将5mg样品与200mg溴化钾混合均匀,压片后测红外光谱,光谱测试范围4000-500cm-1。dsc测试采用美国perkin–elmer公司dsc8500型差示扫描量热仪,测试温度及热焓用高纯标准样品铟校准;精确称量5mg左右样品,气氛为n2(流量为20ml/min),升温和降温速率为10℃/min,扫描范围从-20~120℃。sem测试采用日本电子株式会社jsm-7500f型扫描电子显微镜观察样品的形貌,样品进行喷金处理。锥形量热测试采用英国firetesttechnology公司的锥型量热仪,选用iso5660-1标准,将10g微胶囊与40g环氧树脂混合固化,样品尺寸:100mm×100mm×10mm。极限氧指数测试采用atlaslimitingoxygenindex仪,液化石油气为点燃火源,将10g微胶囊与40g环氧树脂混合固化,样品尺寸:125mm×10mm×4mm。实施例1本实施例中,制备含磷丙烯酸单体的步骤如下:将10moln,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于45kg二氯甲烷后,加入10mol丙烯酰氯、10mol三乙胺,然后在30℃搅拌反应12h,反应时间到达后,将反应液过滤并收集滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的丙烯酰氯和三乙胺,所述饱和氯化钠溶液与滤液的质量比为1:1,再通过减压蒸馏除去萃取所得油相中的溶剂二氯甲烷后得到含磷丙烯酸单体,其结构式如下:本实施例中,制备阻燃型相变微胶囊的步骤如下:(1)在30℃条件下将本实施例制备的1kg含磷丙烯酸单体与10kg甲基丙烯酸甲酯、0.11kg偶氮二异丁腈、11kg十八烷(熔点为28℃)搅拌混合均匀得到油相,在室温下将110kg去离子水和0.22kg十二烷基苯磺酸钠搅拌混合均匀得到水相;(2)在搅拌下于30℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在2000r/min的转速条件下搅拌40min,再于75℃以200r/min的转速搅拌反应4h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到阻燃型相变微胶囊。本实施例制备的阻燃型相变微胶囊呈白色粉状固体,红外谱图如图1所示。图1谱图上的吸收峰:2916cm-1处为c-h的伸缩振动峰,1730cm-1和1149cm-1处为酯键的伸缩振动峰,1237cm-1和1050-980cm-1处的吸收峰分别是由p=o和p-o-c基团产生的;上述吸收峰表明,本实施例通过聚合反应成功制备出了阻燃型相变微胶囊。经dsc测试,本实施例所制备的阻燃型相变微胶囊在升温过程中相转变区间为25~40℃,降温过程中相转变区间为5~25℃(见图2),相变焓为112.3j/g,表明本实施例制备的阻燃型相变微胶囊具有优异的储能能力。由图3可知,本施例制备的阻燃型相变微胶囊为分散良好的微球,粒径均匀平均粒径为120~150nm。对比例1对比例1的步骤如下:(1)在30℃条件下将11kg甲基丙烯酸甲酯、0.11kg偶氮二异丁腈、11kg十八烷搅拌混合均匀得到油相;在室温下将110kg去离子水和0.22kg十二烷基苯磺酸钠搅拌混合,得到水相;(2)在搅拌下于30℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在2000r/min的转速条件下搅拌40min,再于75℃以200r/min的转速搅拌反应4h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到普通相变微胶囊。经dsc测试,本对比例所制备的普通相变微胶囊在升温过程中相转变区间为25~40℃,降温过程中相转变区间为5~25℃,相变焓为112.9j/g。为了比较实施例1制备的相变微胶囊与对比例1制备的相变微胶囊的阻燃性能,将两种相变微胶囊进行了锥形量热测试和极限氧指数测试,测试结果见表1。表1锥形量热测试和极限氧指数测试数据从表1可以看出,实施例1制备的相变微胶囊,其最大释热速率、总释放热、总释烟量显著低于对比例1制备的相变微胶囊,其残余质量显著高于对比例1制备的相变微胶囊,其极限氧指数也高于对比例1制备的相变微胶囊,因此实施例1制备的相变微胶囊与对比例1制备的相变微胶囊相比,阻燃性能大幅度提高。实施例2本实施例中,制备含磷丙烯酸单体的步骤如下:将10moln,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于40kg二氯乙烷后,加入30mol丙烯酰氯、30mol三乙胺,然后在10℃搅拌反应24h,反应时间到达后,将反应液过滤并收集滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的丙烯酰氯和三乙胺,所述饱和氯化钠溶液与滤液的质量比为3:1,再通过减压蒸馏除去萃取所得油相中的溶剂二氯乙烷后得到含磷丙烯酸单体,其结构式如下:本实施例中,制备阻燃型相变微胶囊的步骤如下:(1)在30℃条件下将本实施例制备的0.5kg含磷丙烯酸单体与10kg苯乙烯、0.011kg过氧化二苯甲酰、21kg十六烷(熔点为18℃)搅拌混合均匀得到油相,在室温下将95kg去离子水和0.16kg十二烷基磺酸钠搅拌混合均匀得到水相;(2)在搅拌下于30℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在1000r/min的转速条件下搅拌60min,再于70℃以800r/min的转速搅拌反应8h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到阻燃型相变微胶囊。本实施例制备的阻燃型相变微胶囊呈白色粉状固体,经dsc测试,在升温过程中相转变区间为25~40℃,降温过程中相转变区间为5~25℃(见图2),相变焓达142.7j/g。由图3可知可知,本实施例制备的阻燃型相变微胶囊为分散良好的微球,粒径均匀平均粒径为120~150nm。实施例3本实施例中,制备含磷丙烯酸单体的步骤如下:将10moln,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于65kg三氯甲烷后,加入20mol丙烯酰氯、20mol三乙胺,然后在50℃搅拌反应4h,反应时间到达后,将反应液过滤并收集滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的丙烯酰氯和三乙胺,所述饱和氯化钠溶液与滤液的质量比为2:1,再通过减压蒸馏除去萃取所得油相中的溶剂三氯甲烷后得到含磷丙烯酸单体,其结构式如下:本实施例中,制备阻燃型相变微胶囊的步骤如下:(1)在70℃条件下将本实施例制备的5kg含磷丙烯酸单体与10kg丙烯酸正丁酯、0.45kg偶氮二异庚腈、14kg十八酸(熔点为68℃)搅拌混合均匀得到油相,在室温下将200kg去离子水和0.6kgtween80搅拌混合均匀得到水相;(2)在搅拌下于70℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在3000r/min的转速条件下搅拌10min,再于90℃以500r/min的转速搅拌反应1h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到阻燃型相变微胶囊。实施例4本实施例中,制备含磷丙烯酸单体的步骤如下:将10moln,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于100kg三氯乙烷后,加入15mol丙烯酰氯、15mol三乙胺,然后在20℃搅拌反应18h,反应时间到达后,将反应液过滤并收集滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的丙烯酰氯和三乙胺,所述饱和氯化钠溶液与滤液的质量比为1:1,再通过减压蒸馏除去萃取所得油相中的溶剂三氯乙烷后得到含磷丙烯酸单体,其结构式如下:本实施例中,制备阻燃型相变微胶囊的步骤如下:(1)在60℃条件下将本实施例制备的2kg含磷丙烯酸单体与5kg甲基丙烯酸甲酯、5kg苯乙烯、0.06kg偶氮二异丁腈、8kg十八醇(熔点为58℃)搅拌混合均匀得到油相,在室温下将120kg去离子水和0.4kgspan80搅拌混合均匀得到水相;(2)在搅拌下于60℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在1500r/min的转速条件下搅拌40min,再于80℃以300r/min的转速搅拌反应3h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到阻燃型相变微胶囊。实施例5本实施例中,制备含磷丙烯酸单体的步骤如下:将10moln,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于90kg四氯甲烷后,加入25mol甲基丙烯酰氯、3.5kg质量浓度25~28%的氨水(25~28mol),然后在40℃搅拌反应10h,反应时间到达后,将反应液过滤并收集滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的甲基丙烯酰氯和氨水,所述饱和氯化钠溶液与滤液的质量比为1:1,再通过减压蒸馏除去萃取所得油相中的溶剂四氯甲烷后得到含磷丙烯酸单体,其结构式如下:本实施例中,制备阻燃型相变微胶囊的步骤如下:(1)在25℃条件下将本实施例制备的1kg含磷丙烯酸单体与8kg甲基苯乙烯、2kg甲基丙烯酸、0.11kg过氧化二苯甲酰、8kg十四烷(熔点为6℃)、8kg十六烷(熔点为18℃)搅拌混合均匀得到油相,在室温下将200kg去离子水和1kgtween40搅拌混合均匀得到水相;(2)在搅拌下于25℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在2000r/min的转速条件下搅拌30min,再于75℃以600r/min的转速搅拌反应4h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到阻燃型相变微胶囊。实施例6本实施例中,制备含磷丙烯酸单体的步骤如下:将10moln,n-双(2-羟乙基)氨基亚甲基膦酸二乙酯溶于60kg四氯乙烷后,加入10mol2-丁烯酰氯、20mol氢氧化钠,然后在30℃搅拌反应16h,反应时间到达后,将反应液过滤并收集滤液,在室温下用饱和氯化钠溶液萃取滤液除去滤液中未反应的2-丁烯酰氯和氢氧化钠,所述饱和氯化钠溶液与滤液的质量比为3:1,再通过减压蒸馏除去萃取所得油相中的溶剂四氯乙烷后得到含磷丙烯酸单体,其结构式如下:本实施例中,制备阻燃型相变微胶囊的步骤如下:(1)在45℃条件下将本实施例制备的3kg含磷丙烯酸单体与6kg苯乙烯、4kg丙烯酸、0.25kg偶氮二异丁腈、5kg十八烷(熔点为28℃)、8kg二十烷(熔点为37℃)搅拌混合均匀得到油相,在室温下将100kg去离子水和0.25kg十二烷基苯磺酸钠搅拌混合均匀得到水相;(2)在搅拌下于45℃将步骤(1)所得油相滴加到步骤(1)所得水相中,油相滴加完后在3000r/min的转速条件下搅拌20min,再于85℃以800r/min的转速搅拌反应2h,反应时间到达后,将所得反应液减压抽滤得滤饼并用去离子水洗涤所得滤饼以去除未反应的单体,然后将洗涤后的滤饼真空干燥(0.08mpa、50℃、8h)得到阻燃型相变微胶囊。为了说明本发明所述阻燃型相变微胶囊的使用稳定性,将实施例1至6和对比例1所制备的相变微胶囊在0~100℃范围内反复升降温200次,用dsc测试相变微胶囊的相变焓并计算其相变焓损失率。为了说明本发明所述阻燃型相变微胶囊的耐水性,将实施例1至6和对比例1所制备的相变微胶囊在30℃去离子水中浸泡10天后取出,用dsc测试相变微胶囊的相变焓并计算其相变焓损失率,其结果见表2。表2相变焓及相变焓损失率编号相变焓(j/g)200次热循环后相变焓损失率(%)浸泡10天后相变焓损失率(%)对比例1112.93.151.54实施例1112.31.590.81实施例2142.72.131.01实施例3101.31.470.79实施例486.71.420.78实施例5131.51.830.87实施例6115.31.720.85从表2可以看出,实施例1至6所制备的阻燃型相变微胶囊,其200次热循环后的相变焓损失率和浸泡10天后的相变焓损失率均明显小于与对比例1制备的普通相变微胶囊,测试结果表明,实施例1至6所制备的阻燃型相变微胶囊与对比例1制备的普通相变微胶囊相比,使用稳定性明显提高。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1