一种羟甲基环丙基乙腈衍生物及其制备和应用方法与流程
本发明涉及一种羟甲基环丙基乙腈衍生物及其制备和应用方法。
背景技术
孟鲁司特钠是一种治疗哮喘的医保甲类用药,也可用于治疗过敏性鼻炎的病人。该药的一般耐受性良好,副作用较轻微,通常不需中止治疗。应用该药总的副作用发生率与安慰剂相似。
化合物
zhiqiangzhao等在《substituted4-morpholinen-arylsulfonamidesasγ-secretaseinhibitors》(eur.j.med.chem.124,36-48,2016.)报道了用lialh4,四氢呋喃做溶剂的条件下将双酯基还原成双醇;us6531494报道了用lialh4,乙醚和四氢呋喃做溶剂的条件下将双酯基还原成双醇;wo2005012324报道了用nabh4,四氢呋喃作溶剂反应16小时,收率只有48%。
还有以下的合方法:
wo2008058118报道了用nabh4,四氢呋喃和丙酮作溶剂的条件下将双酯基还原成双醇;wo9823565报道了用硼烷-四氢呋喃的条件下将双酯基还原成双醇。
类似的合成方法还包括有:
us20150322076报道了用dmso作溶剂和氰化钠在回流条件下反应18小时,得到产物;cn104860845a报道了用dmf作溶剂和氰化钠在四丁基溴化铵和碘化钠存在的条件下反应18小时,得到产物;cn103936703a报道了用dmso作溶剂和氰化钠在碘化钠存在的条件下80℃反应48小时,得到产物;wo9518107报道了用dmf和甲苯作溶剂和氰化钠在碘化钠存在的条件下反应,得到产物。
目前合成孟鲁司特钠中间体
技术实现要素:
本发明的目的在于提供一种羟甲基环丙基乙腈衍生物及其制备和应用方法。
本发明所采取的技术方案是:
一种羟甲基环丙基乙腈衍生物,其结构式为:
这种羟甲基环丙基乙腈衍生物的制备方法,包括以下步骤:
1)将
2)将
3)将
4)将
制备方法的步骤1)中,还原反应的温度为70℃~850℃,反应的时间为5h~7h。
制备方法的步骤1)中,还原反应所用的还原剂为硼氢化钠,硼氢化钠与
制备方法的步骤1)中,还原反应所用的溶剂为叔丁醇,叔丁醇与硼氢化钠的用量比为(15~30)ml:1g。
制备方法的步骤2)中,脱氯反应所用的脱氯剂为镍铝合金,镍铝合金与
制备方法的步骤3)中,亚砜为氯化亚砜,氯化亚砜与
制备方法的步骤4)中,氰化反应的温度为55℃~65℃,反应的时间为15h~20h。
制备方法的步骤4)中,氰化反应所用的氰化试剂为氰化钠,氰化钠与
这种羟甲基环丙基乙腈衍生物在制备孟鲁司特类化合物中的应用。
本发明的有益效果是:
本发明制备羟甲基环丙基乙腈衍生物的原料价格低廉,其方法简便易行,安全有效,收率高,降低了生产成本,易于工业放大,能使得该产品能够进行规模化生产。
具体而言:
本发明制备反应的条件温和可控,还原过程中用叔丁醇做溶剂,安全的nabh4做为还原剂,反应时间较短,后处理方便。氰化反应条件温和(60℃),反应时间适中(18小时),后处理简单(萃取后浓缩除掉dmso,吸附后就能得到合格产品)。
附图说明
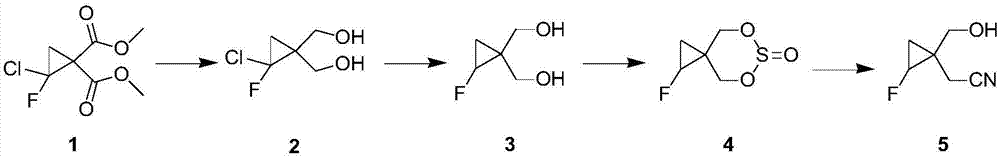
图1是羟甲基环丙基乙腈衍生物的合成线路图;
图2是实施例的具体合成线路图;
图3是实施例所得化合物2的核磁共振氢谱图;
图4是实施例所得化合物4的核磁共振氢谱图;
图5是实施例所得化合物5的核磁共振氢谱图。
具体实施方式
一种羟甲基环丙基乙腈衍生物,其结构式为:
这种羟甲基环丙基乙腈衍生物的合成线路见附图1。下面结合图1中的化合物1~5,进一步说明本发明的制备方法。
这种羟甲基环丙基乙腈衍生物的制备方法,包括以下步骤:
1)将化合物1
2)将化合物2进行脱氯反应,得到化合物3
3)将化合物3与亚砜进行闭环反应,得到化合物4
4)将化合物4进行氰化反应,得到化合物5
优选3的,制备方法的步骤1)4中,还原反应的温度为70℃~80℃,反应的时间为5h~7h;优选的,制备方法的步骤1)中,还原反应的温度为70℃~78℃,反应的时间为5.5h~6.5h。
优选的,制备方法的步骤1)中,还原反应所用的还原剂为硼氢化钠,硼氢化钠与化合物1的摩尔比为(1.5~3):1。
优选的,制备方法的步骤1)中,还原反应所用的溶剂为叔丁醇,叔丁醇与硼氢化钠的用量比为(15~30)ml:1g;进一步优选的,叔丁醇与硼氢化钠的用量比为(18~20)ml:1g。
进一步的,制备方法的步骤1)中,环氧反应具体为:将硼氢化钠和叔丁醇搅拌混合,升温至70℃,滴加化合物1的甲醇溶液,控制温度70℃~78℃搅拌反应6h,反应结束后,降温至50℃,滴加丙酮,继续降温至25℃,滴加浓盐酸,控制温度25℃~35℃,调节ph至3~4,然后分离、洗涤产物,得到化合物2。
优选的,制备方法的步骤1)中,化合物1与甲醇的用量比为1g:(0.5~1)ml;丙酮与化合物1的用量比为(0.2~0.4)g:1g;浓盐酸与化合物1的用量比为(0.8~1.2)g:1g。
优选的,制备方法的步骤2)中,脱氯反应所用的脱氯剂为镍铝合金,镍铝合金与化合物2的摩尔比为(0.5~1):1。
优选的,制备方法的步骤2)中,脱氯反应的温度为70℃~80℃,脱氯反应的时间为2.5h~3.5h。
进一步的,制备方法的步骤2)中,脱氯反应具体为:将化合物2与水混合,加入镍铝合金,升温至70℃,加入乙二胺和氢氧化钠水溶液,控制温度为70℃~80℃,搅拌反应3h,反应结束后,降至室温,加入乙醇,分离产物,然后洗涤提纯,得到化合物3。
优选的,制备方法的步骤2)中,化合物2与水的质量比为(1.5~2.5):1;乙二胺与化合物2的质量比为(0.03~0.05):1;氢氧化钠水溶液浓度为18%~22%,氢氧化钠与化合物2的摩尔比为(2.5~3):1;乙醇与化合物2的用量比为(3~5)ml:1g。
优选的,制备方法的步骤3)中,亚砜为氯化亚砜,氯化亚砜与化合物3的摩尔比为(1~1.5):1。
优选的,制备方法的步骤3)中,闭环反应所用的溶剂为二氯甲烷,二氯甲烷与化合物3的用量比为(8~12)ml:1g。
优选的,制备方法的步骤3)中,闭环反应的温度为-10℃~-5℃。
优选的,制备方法的步骤3)中,闭环反应在氮气或惰性气体保护下进行。
进一步的,制备方法的步骤3)中,闭环反应具体为:将化合物3与二氯甲烷混合搅拌,加入三乙胺,在氮气保护下,控制温度为-10℃~-5℃,加入氯化亚砜进行反应,反应结束后,分离、提纯产物,得到化合物4。
优选的,制备方法的步骤3)中,三乙胺与化合物3的质量比为(1.5~2):1。
优选的,制备方法的步骤4)中,氰化反应的温度为55℃~65℃,反应的时间为15h~20h。
优选的,制备方法的步骤4)中,氰化反应所用的氰化试剂为氰化钠,氰化钠与化合物4的摩尔比为(1~1.5):1。
优选的,制备方法的步骤4)中,氰化反应所用的溶剂为二甲基亚砜,二甲基亚砜与氰化钠的用量比为(4~6)ml:1g。
进一步的,制备方法的步骤4)中,氰化反应具体为:将氰化钠与二甲基亚砜混合,升温至105℃保持1h,然后降至60℃,加入化合物4反应18h,反应完毕后降温至20℃,加入naoh和na2s2o3的水溶液,搅拌30min,加入二氯甲烷萃取,浓缩后蒸馏得到化合物5,化合物5即上述的羟甲基环丙基乙腈衍生物。
这种羟甲基环丙基乙腈衍生物在制备孟鲁司特类化合物中的应用。
优选的,孟鲁司特类化合物为孟鲁司特钠的类似物。
以下通过具体的实施例对本发明的内容作进一步详细的说明。实施例中所用的原料如无特殊说明,均可从常规商业途径得到。
实施例:
第一步:
准备500ml干燥洁净的三口烧瓶,加入叔丁醇(300ml),搅拌下加入硼氢化钠(16.17g,0.427mol),升温至70℃,滴加化合物1(50g,0.237mol)的甲醇(30ml)溶液,控制温度70~78℃(尾气用10%的氢氧化钠溶液吸收后排至大气中),搅拌6小时,tlc监测反应。反应完后,降温至50℃,滴加丙酮(12.4g,0.213mol),继续降温至25℃,滴加浓盐酸(51.4g,0.465mol),控制温度25~35℃,调节ph至3~4。抽滤,乙醇洗涤滤饼(2*50ml),45℃下减压浓缩滤液,加甲醇(100ml),减压浓缩,重复上述操作四次(4*100ml),浓缩物中加甲基叔丁基醚(150ml)搅拌打浆1小时,抽滤,减压浓缩滤液,得黄色油状物化合物237.19g(收率85.56%)。
1h-nmr(400mhz,cdcl3)δ3.92(d,j=11.8hz,1h),3.85(dd,j=19.3,7.2hz,2h),3.75(d,j=11.9hz,1h),3.61(s,2h),1.47(dd,j=16.4,7.8hz,1h),1.20(t,j=7.3hz,1h).
化合物2的核磁共振氢谱图见附图3。
第二步:
准备1000ml干燥洁净的三口烧瓶,加入化合物2(50g,0.324mol),加入水(25g,0.5v),开启搅拌,加入镍铝合金(22.17g,0.259mol),升温至70℃,滴加乙二胺(1.92g,0.032mol),缓慢滴加20%氢氧化钠水溶液(181.4g,0.907mol),控制温度70~80℃,70℃保温搅拌3小时,tlc检测反应进程。反应完后,降至室温,加入乙醇(200ml),搅拌10分钟,抽滤(或沉降后抽取上清液),不能抽干。乙醇洗涤(2*100ml),滤液中加入氯化铵固体(45g)调节ph值至6~7,减压浓缩,加入乙醇(100ml),减压浓缩,加入乙醇(100ml),减压浓缩,加入乙醇(50ml)和甲苯(200ml),减压浓缩干,加入乙醇(50ml),加入甲苯(100ml),减压浓缩干,加入甲基叔丁基醚(150ml),搅拌打浆1小时,抽滤,甲基叔丁基醚洗涤(2*50ml),滤液中加入活性炭(10g),搅拌脱色2小时,垫硅藻土抽滤,甲基叔丁基醚洗涤(2*50ml),减压浓缩滤液,得无色透明油状物化合物3。
1h-nmr(400mhz,cdcl3)δ4.59(ddd,j=64.4,6.1,2.7hz,1h),4.02–3.89(m,1h),3.79–3.70(m,1h),3.58–3.41(m,4h),0.90(ddd,j=9.9,7.3,3.1hz,1h),0.78(dt,j=11.0,6.7hz,1h).
第三步:
准备250ml干燥洁净的三口烧瓶,加入dcm(100ml),搅拌下加入化合物3(10g,0.0833mol),加入三乙胺(18.6g,0.183mol),n2保护,控制温度-10~-5℃,滴加氯化亚砜(10.9g,0.0916mol),tlc监测反应。反应完后,将反应液倒入5%nahco3溶液(100ml)中,搅拌10min,静置分液;再加入dcm(40ml)萃取一次,合并有机相用饱和食盐水洗涤,无水硫酸钠干燥,浓缩至干得化合物4。
化合物4的核磁共振氢谱图见附图4。
第四步:
准备2000ml干燥洁净的三口烧瓶,加入nacn(203g,4.0mol),然后加入dmso(1.0l),开启升温,将内温升至105℃,保持一小时,然后降温至60℃滴加化合物4,保温18小时,反应完全后降温至20℃,滴加naoh和na2s2o3的水溶液,搅拌30min,加入dcm萃取(3.0l×3),浓缩干后蒸馏得化合物5193g(收率74.7%)。化合物5即羟甲基环丙基乙腈衍生物目标产物。
化合物5的核磁共振氢谱图见附图5。
本发明的制备方法大大降低了生产成本,原料价格低廉并操作简单。该方法可以安全放大,使得该产品能够进行规模化生产。
- 还没有人留言评论。精彩留言会获得点赞!