一种3-氧代-2-戊基-环戊烯基乙酸甲酯的制备方法与流程
本发明涉及一种香原料的制备方法,具体涉及一种3-氧代-2-戊基-环戊烯基乙酸甲酯的制备方法。
背景技术:
3-氧代-2-戊基-环戊烯基乙酸甲酯,又名脱氢茉莉酮酸甲酯(dehydrohedione,dhh)是一种重要的香原料,从它出发,通过不对称氢化反应可以制备顺式异构体含量高的高顺二氢茉莉酮酸甲酯。普通的二氢茉莉酮酸甲酯中顺式异构体的含量在10%左右,而高顺二氢茉莉酮酸甲酯中顺式异构体的含量可以高达85%。同普通的二氢茉莉酮酸甲酯相比,高顺二氢茉莉酮酸甲酯香气纯正,强度高,经济价值大,在香精香料行业中得到广泛的使用。
目前文献上报道的3-氧代-2-戊基-环戊烯基乙酸甲酯的制备方法主要有以下几种:
欧洲专利ep0583564和美国专利us6586620报道,从市售普通的二氢茉莉酮酸甲酯出发,经烯醇酯化、环氧化、酸催化重排后可以制得3-氧代-2-戊基-环戊烯基乙酸甲酯。这条路线包括三步反应,操作复杂,而且使用到高浓度的过氧乙酸,具有一定的危险性。其化学反应式如下:
精细化工,2009,26(1),42报道,市售普通的二氢茉莉酮酸甲酯与n-溴代丁二酰亚胺(nbs)或其它的溴化试剂反应,得到α-位取代的溴代二氢茉莉酮酸甲酯,然后在三乙胺或其它碱的催化下脱去溴化氢后制得3-氧代-2-戊基-环戊烯基乙酸甲酯。在溴代反应中因为有两个酮位α-氢可以参与反应,所以这条路线的选择性低,而且反应所用溴代试剂污染大,不适合大规模工业生产。其化学反应式如下:
美国专利申请us20170174607报道,从市售普通的二氢茉莉酮酸甲酯出发,首先制备烯醇甲酯,然后在溴化铜的氧化下得到3-氧代-2-戊基-环戊烯基乙酸甲酯。这条路线需要使用2当量的溴化铜,废料产出大,而且在反应中有部分中间体回复到原料,造成目标产品纯化的困难。其化学反应式如下:
helv.chim.acta.2005,88,3069报道,从市售普通的二氢茉莉酮酸甲酯出发,通过高碘酸氧化脱氢可以制备3-氧代-2-戊基-环戊烯基乙酸甲酯。这条路线得到的粗品含有杂质多,而且高碘酸价格昂贵,不适合大规模工业化生产。其化学反应式如下:
由此可见,现有的制备3-氧代-2-戊基-环戊烯基乙酸甲酯的制备方法存在工艺复杂、成本高、杂质多提纯难度大、污染重不适合工业化生产等问题。
技术实现要素:
本发明的目的在于解决现有技术中存在的问题,提供一种成本低、工艺简单、反应快、产率高、安全环保的3-氧代-2-戊基-环戊烯基乙酸甲酯的制备方法。
以下3-氧代-2-戊基-环戊烯基乙酸甲酯简称dhh,2,3-二氯-5,6-二氰基-1,4-苯醌简称ddq,2,3-二氯-5,6-二氰基-1,4-对苯二酚简称ddhq。
本发明目的是通过以下技术方案来实现的:
一种dhh的制备方法,包括下列步骤:先将二氢茉莉酮酸甲酯溶解在溶剂中,再加入ddq反应后得到终产物粗产品和副产物ddhq,该反应的化学反应式是:
其中以所述二氢茉莉酮酸甲酯为基准量,所述ddq的用量是所述二氢茉莉酮酸甲酯摩尔数的1-1.5倍,所述溶剂的用量是所述二氢茉莉酮酸甲酯摩尔数的8-12倍。
本发明目的还通过以下优选的技术方案来进一步实现:
优选的,对上述反应的产物进行过滤、洗涤、两次浓缩和提纯的后处理。
更优选的,对上述反应的产物进行过滤,用所述溶剂洗涤滤饼后进行第一次浓缩,即去除所述溶剂得到浓缩物,所述浓缩物是终产物粗产品、未反应的ddq和ddhq,再分别向所述浓缩物中加入水和有机溶剂,分层后所述有机溶剂相中主要包含终产物粗产品,对所述有机溶剂相进行第二次浓缩后通过提纯得到终产物。
更优选的,对上述有机溶剂相进行第二次浓缩之前用5%氯化钠水溶液或5%碳酸氢钠水溶液洗涤多次,所述提纯采用的是柱层析或者蒸馏,所述有机溶剂是乙酸乙酯或己烷或环己烷。
优选的,上述反应在酸的催化下进行,所述酸的用量是所述二氢茉莉酮酸甲酯摩尔数的0.05-0.2倍。
优选的,所述酸是盐酸、硫酸、磷酸中任一种无机酸或乙酸、对甲苯磺酸中任一种有机酸。
更优选的,所述酸的用量是所述二氢茉莉酮酸甲酯摩尔数的0.1倍,所述酸是磷酸。
优选的,上述反应中所述ddq的用量是所述二氢茉莉酮酸甲酯摩尔数的1.2倍,所述溶剂的用量是所述二氢茉莉酮酸甲酯摩尔数的10倍。
优选的,上述反应中所述溶剂选自1,4-二氧六环、甲苯、二甲苯中任一种。
更优选的,上述反应中所述溶剂选自1,4-二氧六环。
优选的,所述反应是需要先升温至101-130℃回流再降至室温,其中所述反应时长是4-12h。
与现有技术相比,本发明的有益效果主要体现在:
(1)、虽然二氢茉莉酮酸甲酯和ddq都是公知的,但如同背景技术中提到的那样,现有技术要么是两步反应要么存在高污染,而本发明的dhh的制备方法,创造性地选择从市售普通的二氢茉莉酮酸甲酯出发,在ddq的作用下脱氢一步制得。这条合成制备路线只有一步反应,操作简单,降低了生产的时间成本,是现有技术同类产品中没有采用过的制备方法。
(2)、本发明的dhh的制备方法,在酸催化的条件下反应速度加快,进一步地降低了生产的时间成本。
(3)、本发明使用的原料ddq毒性低,而且副产物ddhq经氧化处理后可以转变回ddq,从而实现重复利用,降低了废物的排放,符合现代绿色化工工艺的基本要素和发展趋势。
(4)、本发明的合成路线避免了现有同类合成方法中所使用的n-溴代丁二酰亚胺(nbs),过氧乙酸,高碘酸等危险、污染大的试剂,操作简单、安全性高,易于工业化放大生产。
(5)、本发明的对有机溶剂相进行第二次浓缩之前用5%氯化钠水溶液或5%碳酸氢钠水溶液洗涤多次是为了进一步降低有机溶剂相中的杂质含量,分层后,有机溶剂相中主要是终产物粗产品,因为终产物粗产品可以很好地溶解在有机溶剂中,而ddq和ddhq这些杂质会富集在水相中,便于后续操作,从而进一步地提高了终产物的纯度。
附图说明
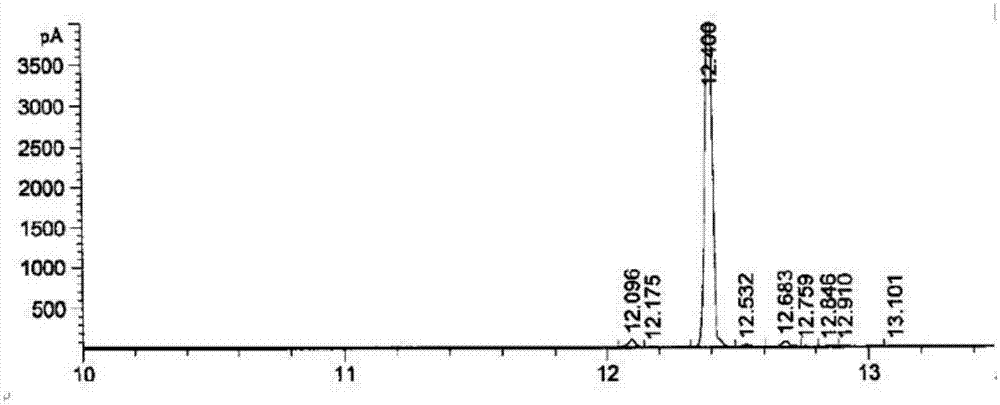
图1为本发明的3-氧代-2-戊基-环戊烯基乙酸甲酯的气相色谱表征;
图2为本发明的3-氧代-2-戊基-环戊烯基乙酸甲酯的核磁表征。
具体实施方式
为使本发明的目的、技术方案及优点更加清楚明白,以下结合附图并举实施例,对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
ddq是药物合成中经常使用的一种氧化剂,对甾类化合物的脱氢芳构化非常有效。其中ddq的结构式是
二氢茉莉酮酸甲酯与ddq在酸作催化剂的条件下可以发生反应,生成dhh,而ddq则转化为ddhq,反应路线如下图所示:
而且如果在上述反应中加入酸,在酸催化的条件下反应速度加快能进一步地缩短反应时间,降低生产成本,其中所用的酸选自盐酸、硫酸、磷酸等无机酸和乙酸、对甲苯磺酸等有机酸,尤其是磷酸效果最好,添加酸的摩尔数为二氢茉莉酮酸甲酯摩尔数的0.05-0.2倍,优选0.1倍。其化学反应式如下:
一种dhh的制备方法,包括下列步骤:(1)上述反应中可以看出,先将市售普通的二氢茉莉酮酸甲酯溶解在溶剂中,再加入ddq反应后得到终产物粗产品和副产物ddhq,这条合成制备路线是从市售普通的二氢茉莉酮酸甲酯出发,在ddq的作用下脱氢一步制得。只有一步反应,降低了生产的时间成本,而且反应快,操作简单,产率较高,易于工业化放大生产。这条合成制备路线是以所述二氢茉莉酮酸甲酯为基准量,所述ddq的用量是所述二氢茉莉酮酸甲酯摩尔数的1-1.5倍,最佳用量为1.2倍;所述溶剂的用量是所述二氢茉莉酮酸甲酯摩尔数的8-12倍,优选10倍,所述溶剂选自1,4-二氧六环,甲苯,二甲苯等沸点比较高的溶剂,优选1,4-二氧六环;上述反应是需要先升温至101-130℃回流再降至室温,其中所述反应时长是4-12h。
(2)对步骤(1)的产物进行过滤,用所述溶剂洗涤滤饼后进行第一次浓缩,即去除所述溶剂得到浓缩物,所述浓缩物是终产物粗产品、未反应的ddq和副产物ddhq,再分别向所述浓缩物中加入水和有机溶剂,分层后有机溶剂层中主要是终产物粗产品和少量的ddq和ddhq;最后对有机溶剂相进行第二次浓缩后提纯得到终产物。这里的滤饼主要是固体ddq和ddhq,用所述溶剂洗涤一次的目的是把滤饼吸附的终产物粗产品洗下来。第一次浓缩是为了去除所述反应用溶剂,因为所述溶剂和水混溶,不利于后处理,所以换成和水不混溶的有机溶剂来提取终产物粗产品,所述有机溶剂可以选自乙酸乙酯或己烷或环己烷,它们和水在一起会分层,而有机溶剂对于终产物粗产品的溶解度很好。对有机溶剂相进行第二次浓缩之前用5%氯化钠水溶液或5%碳酸氢钠水溶液洗涤多次是为了进一步降低有机溶剂相中的杂质含量,分层后,有机溶剂相中主要是终产物粗产品,因为终产物粗产品可以很好地溶解在有机溶剂中,便于后续操作,从而进一步地提高了终产物的纯度。
本发明的制备合成路线使用的原料ddq毒性低,而且副产物ddhq经氧化处理后可以转变回ddq,从而实现重复利用,降低了废物的排放,符合现代绿色化工工艺的基本要素和发展趋势;而且避免了现有同类合成方法中所使用的n-溴代丁二酰亚胺(nbs),过氧乙酸,高碘酸等危险、污染大的试剂,操作简单、安全性高,易于工业化放大生产。
实施例1:
向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),升温至回流(温度为101℃)反应12小时。降至室温后过滤除去固体(主要成分是未反应的ddq以及ddq被还原生成的ddhq),用1,4-二氧六环20ml洗涤滤饼一次。除去溶剂1,4-二氧六环后,向浓缩物中加入150ml水,然后加100ml乙酸乙酯提取一次。乙酸乙酯层用5%氯化钠水溶液洗涤2次,每次50ml。乙酸乙酯层浓缩后得到终产物粗产品15.8克,硅胶柱提纯后得到终产物dhh10.2克,终产物为无色油状物,如图1所示,气相色谱检查的纯度为97.2%,反应产率68%,如图2所示,经核磁表征确定结构正确。
实施例2:
以实施例1为基础,实施例2与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq15.0g(0.066mol),加入市售85%磷酸0.74克(0.007mol)。升温至回流(温度为101℃)反应8小时。得到终产物dhh7.8克,气相色谱检查的纯度为96.3%,反应产率为52.3%,经核磁表征确定结构正确。
实施例3:
向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq22.3g(0.1mol),加入市售85%磷酸0.74克(0.007mol)。升温至回流(温度为101℃)反应5小时。降至室温后过滤除去固体(主要成分是未反应的ddq以及ddq被还原生成的ddhq),用1,4-二氧六环20ml洗涤滤饼一次。除去溶剂1,4-二氧六环后,向浓缩物中加入150ml水,然后加100ml己烷提取一次。己烷层用5%碳酸氢钠水溶液洗涤2次,每次50ml。己烷层浓缩、再用硅胶柱提纯后得到终产物dhh8.3克,终产物为无色油状物,经气相色谱检查的纯度为96.1%,反应产率为55.3%,经核磁表征确定结构正确。
实施例4:
以实施例3为基础,实施例4与实施例3的区别仅在于:向500ml三口烧瓶内加入120ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售85%磷酸0.74克(0.007mol)。升温至回流(温度为101℃)反应5小时。得到终产物dhh9.7克,气相色谱检查的纯度为95.3%,反应产率为64.7%,经核磁表征确定结构正确。
实施例5:
向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售85%磷酸0.74克(0.007mol)。升温至回流(温度为101℃)反应5小时。降至室温后过滤除去固体(主要成分是未反应的ddq以及ddq被还原生成的ddhq),用1,4-二氧六环20ml洗涤滤饼一次。除去溶剂1,4-二氧六环后,向浓缩物中加入150ml水,然后加100ml环己烷提取一次。环己烷层用5%碳酸氢钠水溶液洗涤2次,每次50ml。环己烷层浓缩、再用硅胶柱提纯后得到终产物dhh10.7克,终产物为无色油状物,经气相色谱检查的纯度为95.3%,反应产率为71.3%,经核磁表征确定结构正确。
实施例6:
以实施例5为基础,实施例6与实施例5的区别仅在于:向500ml三口烧瓶内加入180ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售85%磷酸0.74克(0.007mol)。升温至回流(温度为101℃)反应5小时。得到终产物dhh10.4克,气相色谱检查的纯度为96.1%,反应产率为69.2%,经核磁表征确定结构正确。
实施例7:
以实施例1为基础,实施例7与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售37%盐酸0.7克(0.007mol)。升温至回流(温度为101℃)反应4小时。得到终产物dhh7.23克,气相色谱检查的纯度为96.9%,反应产率为48.3%,经核磁表征确定结构正确。
实施例8:
以实施例1为基础,实施例8与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售浓硫酸0.69克(0.007mol)。升温至回流(温度为101℃)反应4小时。得到终产物dhh8.3克,气相色谱检查的纯度为95.1%,反应产率为55.3%,经核磁表征确定结构正确。
实施例9:
以实施例1为基础,实施例9与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入对甲苯磺酸2.4克(0.014mol)。升温至回流(温度为101℃)反应9小时。得到终产物dhh9.2克,气相色谱检查的纯度为95.7%,反应产率为61.3%,经核磁表征确定结构正确。
实施例10:
以实施例1为基础,实施例10与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),和0.42克的乙酸(0.007mol)。升温至回流(温度为101℃)反应8小时。得到终产物dhh9.3克,气相色谱检查的纯度为96.7%,反应产率为62%,经核磁表征确定结构正确。
实施例11:
向500ml三口烧瓶内加入150ml甲苯,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),和0.42克(0.007mol)的乙酸。升温至回流(温度为110℃)反应8小时。降至室温后过滤除去固体(ddq和ddq被还原生成的ddhq),用20ml甲苯洗涤滤饼一次。除去溶剂甲苯后,向浓缩物中加入150ml水,然后加100ml乙酸乙酯提取一次。乙酸乙酯层用5%氯化钠水溶液洗涤2次,每次50ml。乙酸乙酯层浓缩后得到终产物粗产品16.8克,硅胶柱提纯后得到终产物dhh7.7克,终产物为无色油状物,气相色谱检查的纯度为95.1%,反应产率为51.2%,经核磁表征确定结构正确。
实施例12:
以实施例11为基础,实施例12与实施例11的区别仅在于:向500ml三口烧瓶内加入150ml甲苯,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),和市售85%磷酸0.74克(0.007mol)。升温至回流(温度为110℃)反应8小时。得到终产物dhh8.5克,气相色谱检查的纯度为97.0%,反应产率为56.7%,经核磁表征确定结构正确。
实施例13:
向500ml三口烧瓶内加入150ml二甲苯,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),和市售85%磷酸0.74克(0.007mol)。升温至回流(温度为130℃)反应8小时。降至室温后过滤除去固体(ddq和ddq被还原生成的ddhq),用20ml二甲苯洗涤滤饼一次。除去溶剂二甲苯后,向浓缩物中加入150ml水,然后加100ml乙酸乙酯提取一次。乙酸乙酯层用5%氯化钠水溶液洗涤2次,每次50ml。乙酸乙酯层浓缩后得到终产物粗产品17.2克,硅胶柱提纯后得到终产物dhh9.2克,终产物为无色油状物,气相色谱检查的纯度为97.0%,反应产率为61.3%,经核磁表征确定结构正确。
实施例14:
以实施例1为基础,实施例14与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售85%磷酸0.37克(0.004mol)。升温至回流(温度为101℃)反应12小时。得到终产物dhh9.8克,气相色谱检查的纯度为95.3%,反应产率为65.3%,经核磁表征确定结构正确。
实施例15:
以实施例1为基础,实施例15与实施例1的区别仅在于:向500ml三口烧瓶内加入150ml1,4-二氧六环,15g(0.066mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq18.1g(0.08mol),加入市售85%磷酸1.48克(0.014mol)。升温至回流(温度为101℃)反应5小时。得到终产物dhh6.2克,气相色谱检查的纯度为95.3%,反应产率为41.3%,经核磁表征确定结构正确。
实施例16:
向3000ml三口烧瓶内加入1500ml1,4-二氧六环,150g(0.66mol)市售二氢茉莉酮酸甲酯,搅拌溶解后加入ddq181g(0.8mol),和7.5克(0.066mol)的市售85%磷酸。升温至回流(温度为101℃)反应5.5小时。降至室温后过滤除去固体(ddq和ddq被还原生成的酚),用1,4-二氧六环200ml洗涤滤饼一次。除去溶剂1,4-二氧六环后,向浓缩物中加入1500ml水,然后加1000ml乙酸乙酯提取一次。乙酸乙酯层用5%氯化钠水溶液洗涤2次,每次250ml;用5%碳酸氢钠水溶液洗涤一次,用量250ml。乙酸乙酯层浓缩后得到终产物粗产品160克,快速蒸馏后再精馏提纯,得到终产物dhh94克,终产物为无色油状物,气相色谱检查的纯度为95.7%,反应产率为62.7%,经核磁表征确定结构正确。
以上所述仅为本发明的较佳的实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!