一种从茶花粉中分离纯化咖啡因的方法与流程
本发明涉及分离提纯技术领域,具体涉及一种从茶花粉中分离纯化咖啡因的方法。
背景技术:
茶花粉是雄性生殖细胞,是遗传基因源,担负着植物遗传的任务,具有全面的营养及多种生物学功能。其活性成分如多酚、黄酮、多糖以及脂质类物质等对茶花粉的抗氧化、抑菌、降血糖、降血脂、抗肿瘤、调节机体免疫能力等生物活性具有极其重要的作用。
咖啡因是从茶叶、咖啡果中提取出来的一种生物碱,具有预防阿尔茨海默病等神经疾病、扩张血管、保护肝损伤等生理功能。目前,咖啡因主要是通过大孔树脂吸附层析法、升华法、萃取法等手段从茶叶中分离纯化出。但是,上述的分离纯化方法存在分离时间长、有机试剂使用较多、对环境造成污染、纯度不高等缺点,因此在咖啡因的分离纯化方面,新的替代方法越来越受关注。
技术实现要素:
本发明旨在至少在一定程度上解决上述技术中的技术问题之一,即分离纯化的咖啡因具有纯度高,杂质少的优点。为此,本发明的一个目的在于提出一种从茶花粉中分离纯化咖啡因的方法。
为达到上述目的,本发明实施例提出了一种从茶花粉中分离纯化咖啡因的方法,其包括以下步骤:
s1、提取:取茶花粉,加入乙醇,于水浴中浸提,共提取3次,抽滤,合并滤液,并减压浓缩成浸膏,得粗提物;
s2、萃取:粗提物加入蒸馏水,搅拌溶解,依次用石油醚、乙酸乙酯和正丁醇萃取,均萃取3次,而后将它们分别减压浓缩,得石油醚相、乙酸乙酯相及正丁醇相;
s3、大孔吸附树脂层析:取乙酸乙酯相,加入蒸馏水复溶,振荡离心12min,取上清液加入大孔树脂中,大孔树脂置于摇床中吸附16h,将吸附后的大孔树脂装柱,使用不同浓度的乙醇水溶液进行洗脱,洗脱体积均为5个柱体积,收集各洗脱组分,获得大孔树脂乙醇洗脱相;
s4、酸解:取大孔树脂乙醇洗脱相,加入无水乙醇,而后再加入等体积4m的盐酸,于90℃水浴中酸水解90min,将酸解液减压蒸干,获得大孔树脂乙醇洗脱相酸解物;
s5、hsccc制备分离:配置正己烷-乙酸乙酯-甲醇-水溶液体系,体积比为4:6:4:6,充分震荡,静置分层,分别收集上相和下相溶液,超声脱气,上相为固定相,下相为流动相,将固定相按照一定的流速泵满色谱柱后,调节高速逆流色谱转速至850rpm,并将流动相按照一定的流速泵入色谱柱中;两相达平衡后,将样品溶液从进样环注入高速逆流色谱管路,样品溶液为步骤s4获得的大孔树脂乙醇洗脱相酸解物,同时通过紫外检测器检测流出组分,紫外检测波长设置为280nm,根据峰形收集各组分;
s6、半制备型高效液相纯化:将步骤s5收集到的组分按顺序编号,以咖啡因标准品作为对照,进行高效液相色谱分析,选择与咖啡因标准品峰保留时间一致的样品作为研究对象进行半制备型高效液相纯化,其上样条件为:质量浓度为5mg/ml-20mg/ml,进样体积为10μl-200μl,根据峰形收集各组分,进行高效液相色谱分析,筛选出目标组分;
s7、结构鉴定:目标组分利用高效液相色谱、核磁共振、质谱的方法进行鉴定,即可确定目标组分为咖啡因;
s8、纯度测定:将s7中的目标组分利用高效液相色谱进行测定,按照峰面积归一法确定其纯度。
根据本发明实施例的一种从茶花粉中分离纯化咖啡因的方法,其以茶花粉为原料,通过提取、萃取、大孔吸附树脂层析和酸解处理后,再采用高速逆流色谱(hsccc)与半制备型高效液相纯化获得化合物,最后通过高效液相色谱、核磁共振、质谱进行鉴定确定所纯得化合物为咖啡因,从而实现了从茶花粉中直接分离纯化出咖啡因,所得到的咖啡因纯度高、杂质少。
另外,根据本发明上述实施例提出的一种从茶花粉中分离纯化咖啡因的方法,还可以具有如下附加的技术特征:
根据本发明实施例,所述步骤s1中,茶花粉与乙醇的料液比1g:10ml;步骤s2中,粗提物与蒸馏水的料液比1g:10ml;步骤s3中,乙酸乙酯与蒸馏水的料液比1g:10ml;步骤s4中,大孔树脂乙醇洗脱相与无水乙醇的料液比0.1g:50ml。
根据本发明实施例,所述步骤s1中,茶花粉浸提时乙醇浓度为80%。
根据本发明实施例,所述步骤s3中不同浓度的乙醇水溶液进行洗脱,其洗脱条件为:依次用浓度0%、10%、30%的乙醇水溶液进行洗脱,获得大孔树脂30%乙醇洗脱相。
根据本发明实施例,所述步骤s1中,浸提时,在80℃水浴条件下浸提6h。
根据本发明实施例,步骤s5中,固定相以30ml/min的流速泵入色谱柱,流动相以3ml/min的流速泵入色谱柱。
根据本发明实施例,所述步骤s6中,样品质量浓度为5mg/ml。
根据本发明实施例,所述步骤s6中,进样量为200μl。
附图说明
图1是根据本发明实施例一hsccc分离色谱图;
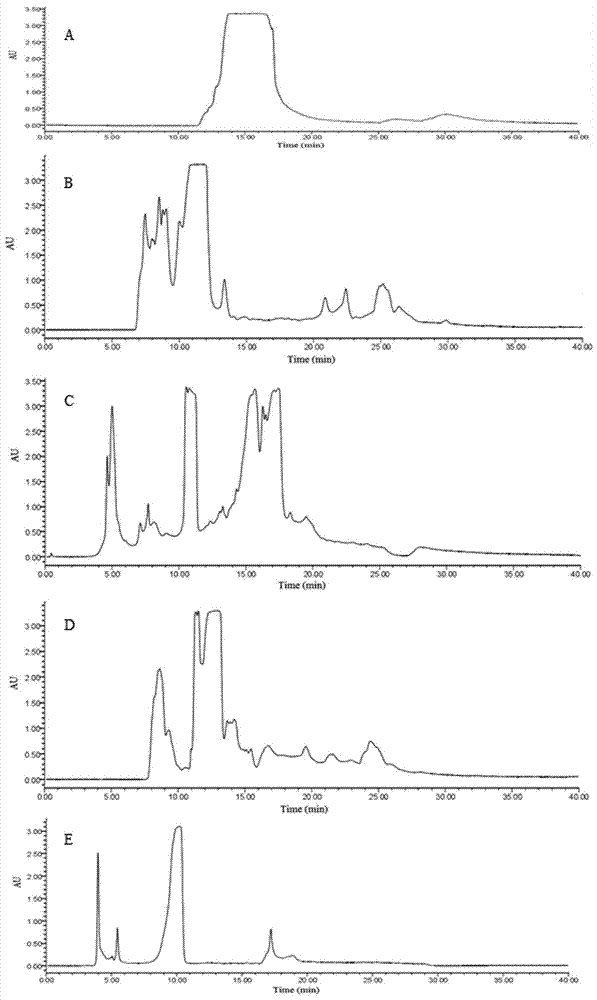
图2是根据本发明实施例一分离样品peaki-peakiv的高效液相色谱图;
图3是根据本发明实施例一咖啡因标准品的高效液相色谱图;
图4是根据本发明实施例一分离样品peakii进样质量浓度的优化;
图5是根据本发明实施例二分离样品peakii进样量的优化;
图6是根据本发明实施例三分离样品compoundi的高效液相图;
图7是根据本发明实施例三分离样品compoundi的核磁共振图;
图8是根据本发明实施例三咖啡因标准品的核磁共振图;
图9是根据本发明实施例三分离样品compoundi的质谱图。
具体实施方式
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
本发明提供一种从茶花粉中分离纯化咖啡因的方法,其主要解决的是现有的在分离纯化咖啡因的方法存在着分离时间长、有机试剂使用较多、对环境造成污染、纯度不高等缺点;为了解决上述问题,该方法采用的方案是:其以茶花粉为原料,通过提取、萃取、大孔吸附树脂层析和酸解处理后,再采用高速逆流色谱(hsccc)与半制备型高效液相纯化获得化合物,最后通过高效液相色谱、核磁共振、质谱进行鉴定确定所纯得化合物为咖啡因,从而实现了从茶花粉中直接分离纯化出咖啡因,所得到的咖啡因纯度高、杂质少。
为了更好的理解上述技术方案,下面将参照附图更详细地描述本发明的示例性实施例。虽然附图中显示了本发明的示例性实施例,然而应当理解,可以以各种形式实现本发明而不应被这里阐述的实施例所限制。相反,提供这些实施例是为了能够更透彻地理解本发明,并且能够将本发明的范围完整的传达给本领域的技术人员。
为了更好的理解上述技术方案,下面将结合说明书附图1-9以及具体的实施方式对上述技术方案进行详细的说明。
实施例一进样质量浓度的影响
s1、提取
准确称取1000g经高速万能粉碎机粉碎后,过筛(40目)后的茶花粉,按料液比1g:10ml的比例添加80%乙醇,于80℃水浴条件下浸提6h,共提取3次,抽滤获得上清液,合并上清液,并于50℃下减压浓缩成浸膏,获得粗提物,4℃冷藏备用。
s2、萃取
将步骤s1获得的粗提物按料液比1g:10ml加入蒸馏水,搅拌溶解,依次用石油醚、乙酸乙酯和正丁醇萃取,均萃取3次,而后将它们分别置于50℃旋转蒸发仪中减压浓缩,获得石油醚相、乙酸乙酯相及正丁醇相。
s3、大孔吸附树脂层析
取步骤s2中的乙酸乙酯相,按料液比1g:10ml加入蒸馏水复溶,振荡,4000r/min离心12min,取上清液加入hp-20型大孔树脂,然后将大孔树脂置于37℃下150r/min的摇床中吸附16h,将吸附后的大孔树脂装柱,依次用浓度为0%、10%、30%的乙醇水溶液进行洗脱,洗脱体积均为5个柱体积,然后收集各洗脱组分,得大孔树脂30%乙醇洗脱相,4℃冷藏备用。
s4、酸解
取步骤s3获得的大孔树脂30%乙醇洗脱相,按料液比0.1g:50ml的比例加入无水乙醇,而后再加入等体积4m的盐酸,于90℃水浴中酸水解90min,酸水解后,将酸解液置于50℃旋转蒸发仪上减压蒸干,获得大孔树脂30%乙醇洗脱相酸解物,即hfr3。
s5、hsccc制备分离
配置正己烷-乙酸乙酯-甲醇-水溶液体系,按照正己烷:乙酸乙酯:甲醇:水的体积比为4:6:4:6的比例量取各溶剂于分液漏斗中,充分震荡,静置分层,分别收集上相和下相溶液,超声脱气30min,以上相作为hsccc的固定相,下相作为hsccc的流动相;采用反向模式,即从头到尾进行洗脱,首先将上相溶液(固定相)以30ml/min的流速泵满色谱柱后,将转速调至850rpm,并将下相溶液(流动相)以3ml/min的流速泵入色谱柱中;两相达平衡后(主机螺旋管内流动相与固定相达到动力学平衡,即检测器端口有流动相被置换出时),将样品溶液从进样环注入高速逆流色谱管路,样品溶液为大孔树脂30%乙醇洗脱相酸解物,同时通过紫外检测器检测流出组分,紫外检测波长设置为280nm,根据峰形手动收集各组分,分离出四个组分,如图1所示。
s6、半制备型高效液相纯化
将步骤s5收集到的四个组分按顺序编号peaki、peakii、peakiii、peakiv。然后以咖啡因标准品作为对照,利用高效液相色谱对峰组分peaki、peakii、peakiii、peakiv和咖啡因标准品进行分析,结果如图2和图3所示,通过对比分析,只有peakii含有与咖啡因标准品保留时间一致的色谱峰,初步确定样品peakii可能为咖啡因;选择与咖啡因标准品峰保留时间一致的样品作为研究对象用半制备型高效液相色谱纯化,其上样条件为:质量浓度5mg/ml-20mg/ml,进样体积200μl,根据峰形收集目标组分,进行高效液相色谱分析,结果如图4所示。
如图4所示,实验分别考察了peakii进样质量浓度为5mg/ml、8mg/ml、10mg/ml、15mg/ml、20mg/ml时的分离效果,其液相图如图4所示。由图4a可知,当peakii进样质量浓度为5mg/ml时,咖啡因峰形对称,有利于收集。由图4b可知,当进样质量浓度增加到8mg/ml时,物质的分离度下降,出现了明显的过载现象。综合考虑,选择最佳进样质量浓度为5mg/ml。
实施例二进样体积的影响
该实施例从茶花粉中分离纯化咖啡因的方法的原理和步骤基本上与实施例一相同,相同的地方不再详细描述,仅描述不同的地方,不一样的在于:其中步骤s6中,选择peakii作为研究对象用半制备型高效液相色谱纯化,其上样条件为:质量浓度5mg/ml,进样体积5μl-200μl,根据峰形收集目标组分,进行高效液相色谱分析。
如图5所示,实验分别考察了peakii进样体积为5μl、50μl、100μl、150μl、200μl时的分离效果,其液相图如图5所示。在确定了peakii进样质量浓度为5mg/ml时,进样体积成为影响制备效能的重要因素,不仅影响分离度,同时也决定了制备效率和产物纯度。由图5c可知,当进样体积为100μl时,咖啡因峰形对称,分离度高,有利于进行收集。随着加大样品peakii的进样体积,由图5e可知,当进样体积为200μl时,咖啡因的峰形没有发生改变,在不影响分离度的情况下,单次进样体积越大越好,节约了实验时间,减少了流动相消耗量,提高了实验效率。综合考虑,选择最佳进样体积为200μl。
实施例三分离纯化产物的鉴定
该实施例从茶花粉中分离纯化咖啡因的方法的原理和步骤基本上与实施例一相同,相同的地方不再详细描述,仅描述不同的地方,不一样的在于:其中步骤s6中,选择peakii作为研究对象用半制备型高效液相色谱纯化,其上样条件为:质量浓度5mg/ml,进样体积200μl,根据峰形收集目标组分。
s7、结构鉴定、
将s6收集得到的组分命名为compoundi,再利用高效液相色谱、核磁共振、质谱的方法进行鉴定,即可确定收集的组分为咖啡因。
利用高效液相色谱对compoundi进行分析,结果如图6所示。与图3中咖啡因标准品对比分析可知,compoundi与咖啡因标准品的保留时间一致,初步确定样品compoundi可能为咖啡因。
接着,分别对compoundi与咖啡因标准品进行核磁共振分析,结果如图7和图8所示。图7中,compoundi核磁数据如下:1hnmr(400mhz,cd3od,δ,ppm,j/hz):7.78(1h,s,c8-h),3.87(3h,s,n7-ch3),3.41(3h,s,n3-ch3),3.23(3h,s,n1-ch3)。图8中,咖啡因标准品的核磁数据为:1hnmr(400mhz,cd3od,δ,ppm,j/hz):7.87(1h,s,c8-h),3.99(3h,s,n7-ch3),3.54(3h,s,n3-ch3),3.35(3h,s,n1-ch3)。通过对比分析,可知咖啡因和样品compoundi的核磁数据一致。因此确定样品compoundi为咖啡因。
最后,对样品compoundi进行质谱分析,结果如图9所示,从图中可以看到m/z
195[m-h]+的离子碎片,确定样品compoundi的相对分子量为194。样品compoundi与咖啡因的相对分子量一致,因此进一步确定样品compoundi为咖啡因。
实施例四分离纯化产物的纯度测定
该实施例从茶花粉中分离纯化咖啡因的方法的原理和步骤基本上与实施例一相同,相同的地方不再详细描述,仅描述不同的地方,不一样的在于:其中步骤s7中,将s7中compoundi鉴定为咖啡因的样品,使用高效液相色谱对其进行纯度分析,确定其纯度。
s8、纯度测定:将s7中的目标组分利用高效液相色谱进行测定,按照峰面积归一法确定其纯度。
使用高效液相色谱对compoundi进行纯度分析,结果如图4所示,使用峰面积归一法测得compoundi纯度为94.05%,即从茶花粉中纯化获得的咖啡因纯度为94.05%。
综上可知,本发明从茶花粉中分离纯化咖啡因的方法,可实现了从茶花粉中直接分离纯化出对咖啡因,所得到的咖啡因纯度高(纯度为94.05%)、杂质少。
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不应理解为必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。此外,本领域的技术人员可以将本说明书中描述的不同实施例或示例进行接合和组合。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 还没有人留言评论。精彩留言会获得点赞!