基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物及其制备方法与流程
本发明涉及共轭微孔聚合物技术领域,具体涉及一种基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物及其制备方法。
背景技术:
随着信息化时代的到来和科学技术的飞速发展,大规模及超大规模集成电路应运而生,其在人们生活中应用十分普及。为匹配集成电路高速发展和人们对高续航需求,需要专业的、高性能、大容量、轻便灵巧、经济耐用、充电速度快的电池材料,而锂电池的发展正处于瓶颈期,因其能量密度已经接近其物理极限,继续开发成本与性能提升比值较低。
有机多孔材料近年来因其独特结构与性能引发科研人员的密切关注,不少科研人员投身于有机多孔材料的开发研究,发现有机多孔材料在气体吸附、分离、非均相催化和储能等领域存在巨大应用价值,进而近年来,外界把寻找超级电池的突破口投向这一新型材料。
有机多孔材料(mops)按照其结构特点分为四种类型:自具微孔聚合物(polymersofintrinsicmicroporosity,pims)、超交联聚合物(hyper-cross-linkedpolymers,hcps)、共价有机网络(covalentorganicframeworks,cofs)和共轭微孔聚合物(conjugatedmicroporouspolymers,cmps)。共轭微孔聚合物(cmps)作为有机多孔材料的一个分支,自cooper课题组初次以2d芳基炔化物、芳基溴/碘化物为单体成功合成3dcmps(paes)后,越来越多的研究者致力于cmps的研究。cmps具有很多优点,如比表面积大、骨架密度低,分子内部含有开放连通的分子尺寸孔道,具有稳定的物理性质和化学性质。
按照不同的化学反应构建结构单元,进而发展不同结构和特殊性质的cmps,成为目前的研究热点。
技术实现要素:
本发明的目的在于提供一种基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物及其制备方法,本发明提供的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物呈现片状结构,为非晶形材料,且具备紫外吸收性能。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物,以1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮为单体、按1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮摩尔比为1:(1.3~1.7)制备得到,具有式i所示结构:
式i中,
所述1,3,5-三(4-醛基吡啶基)三嗪具有式ii所示结构:
本发明提供了上述技术方案所述基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的制备方法,包括以下步骤:
将三聚氯氰、4-吡啶醛和四氢呋喃混合,在保护气氛中进行取代反应,得到1,3,5-三(4-醛基吡啶基)三嗪;
在保护气氛中,将所述1,3,5-三(4-醛基吡啶基)三嗪、对称引达省-1,3,5,7(2h,6h)-四酮、有机溶剂、酸性催化剂和水混合,进行亲核加成-消除反应,得到具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物;其中,所述1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮的摩尔比为1:(1.3~1.7)。
优选地,所述三聚氯氰和4-吡啶醛的摩尔比为1:(2.5~3.5)。
优选地,所述取代反应的温度为65~75℃,时间为70~75h。
优选地,所述酸性催化剂包括甲酸、乙酸和丙酸中的一种或几种。
优选地,所述有机溶剂和水的体积比为(10~20):1。
优选地,所述有机溶剂包括正丁醇、邻二氯苯、均三甲苯和二噁烷中的一种或几种。
优选地,所述亲核加成-消除反应的温度为110~130℃,时间为70~75h。
优选地,所述亲核加成-消除反应完成后还包括:
将亲核加成-消除反应完成后所得体系进行固液分离,依次采用n,n-二甲基甲酰胺和乙醇对所得固体物料进行洗涤,干燥,依次采用甲醇和二氯甲烷对所得干燥物料进行索氏提取,干燥,得到具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物。
本发明提供了一种具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物,本发明在1,3,5-三(4-醛基吡啶基)三嗪基础上引入对称引达省-1,3,5,7(2h,6h)-四酮,制备得到的共轭微孔聚合物呈现片状结构,为非晶形材料,且具备紫外吸收性能。
本发明提供了具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的制备方法,方法简单易行,有利于实现规模化生产。
附图说明
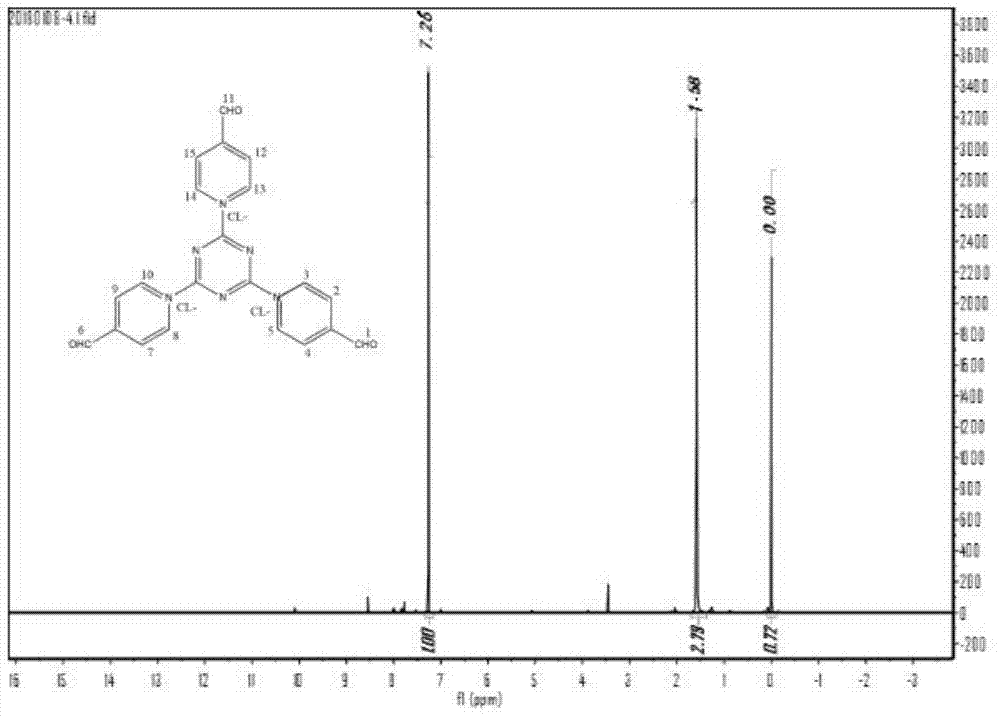
图1为实施例1中1,3,5-三(4-醛基吡啶基)三嗪的核磁氢谱图;
图2为实施例1中1,3,5-三(4-醛基吡啶基)三嗪的红外光谱图;
图3为实施例1中1,3,5-三(4-醛基吡啶基)三嗪的紫外光谱图;
图4为实施例1中对称引达省-1,3,5,7(2h,6h)-四酮的核磁氢谱图;
图5为实施例1中对称引达省-1,3,5,7(2h,6h)-四酮的红外光谱图;
图6为实施例1中具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的红外光谱图;
图7为实施例1中具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的紫外光谱图;
图8为实施例1中具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的热重分析图;
图9为实施例1中具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的xrd扫描分析图;
图10为实施例1中具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的电镜扫描图。
具体实施方式
本发明提供了一种基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物,以1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮为单体、按1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮摩尔比为1:(1.3~1.7)制备得到,具有式i所示结构:
式i中,
所述1,3,5-三(4-醛基吡啶基)三嗪具有式ii所示结构:
本发明提供了上述技术方案所述基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的制备方法,包括以下步骤:
将三聚氯氰、4-吡啶醛和四氢呋喃混合,在保护气氛中进行取代反应,得到1,3,5-三(4-醛基吡啶基)三嗪;
在保护气氛中,将所述1,3,5-三(4-醛基吡啶基)三嗪、对称引达省-1,3,5,7(2h,6h)-四酮、有机溶剂、酸性催化剂和水混合,进行亲核加成-消除反应,得到具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物;其中,所述1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮的摩尔比为1:(1.3~1.7)。
本发明将三聚氯氰、4-吡啶醛和四氢呋喃混合,在保护气氛中进行取代反应,得到1,3,5-三(4-醛基吡啶基)三嗪。在本发明中,所述三聚氯氰和4-吡啶醛的摩尔比优选为1:(2.5~3.5)。本发明对于所述三聚氯氰、4-吡啶醛和四氢呋喃混合没有特殊的限定,采用本领域技术人员熟知的物料混合的技术方案即可;在本发明中,所述混合优选在搅拌条件下进行。在本发明中,所述取代反应的温度优选为65~75℃,更优选为70℃;时间优选为70~75h,更优选为72h。本发明对于提供所述保护气氛的保护气体种类没有特殊的限定,采用本领域技术人员熟知的保护气体即可,具体如氮气。在本发明的实施例中,具体是将三聚氯氰、4-吡啶醛和四氢呋喃混合,搅拌1.5~2.5h,使物料充分混合并初步进行反应,然后在保护气氛中、65~75℃条件下反应70~75h。
完成所述取代反应后,本发明优选将所得体系进行固液分离,依次采用四氢呋喃和乙醇对所得固体物料进行洗涤,干燥,得到1,3,5-三(4-醛基吡啶基)三嗪。本发明对于所述固液分离的方式没有特殊的限定,采用本领域技术人员熟知的固液分离的技术方案即可,具体如减压抽滤。本发明对于所述干燥没有特殊的限定,采用本领域技术人员熟知的干燥的技术方案、能够将物料充分干燥即可。
得到1,3,5-三(4-醛基吡啶基)三嗪后,本发明在保护气氛中,将所述1,3,5-三(4-醛基吡啶基)三嗪、对称引达省-1,3,5,7(2h,6h)-四酮、有机溶剂、酸性催化剂和水混合,进行亲核加成-消除反应,得到具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物;其中,所述1,3,5-三(4-醛基吡啶基)三嗪和对称引达省-1,3,5,7(2h,6h)-四酮的摩尔比为1:(1.3~1.7)。在本发明中,所述酸性催化剂优选包括甲酸、乙酸和丙酸中的一种或几种。
在本发明中,所述有机溶剂和水的体积比优选为(10~20):1,更优选为(13~17):1。在本发明中,所述有机溶剂能够增加反应原料的溶解度,所述水能够调节反应体系的极性,保证反应顺利进行。
在本发明中,所述有机溶剂优选包括正丁醇、邻二氯苯、均三甲苯和二噁烷中的一种或几种。
本发明对所述对称引达省-1,3,5,7(2h,6h)-四酮的来源没有特殊的限定,采用本领域技术人员熟知的方法制备得到即可。在本发明的实施例中,所述对称引达省-1,3,5,7(2h,6h)-四酮的制备方法具体包括以下步骤:
将均苯四甲酸酐、乙酰乙酸乙酯和三乙胺在搅拌条件下混合,水浴加热至60~65℃,向所得混合物中加入乙酸酐,油浴加热至98~102℃回流2~2.5h;反应结束后冷却至室温,然后在0~5℃条件下冷却12~15h,出现淡褐色沉淀物,将所得物料减压抽滤,分别用乙酸酐和乙醚洗涤,得到橙色沉淀物,干燥;将干燥后所得产物按1:100比例加入蒸馏水溶解,在冰水浴条件下,向所得物料中滴加浓硫酸,待溶液变咖啡色,再继续滴加1~2ml浓硫酸,将所得物料用乙醇进行洗涤,干燥;将干燥后所得产物与120ml乙腈混合,在搅拌、在90~95℃条件下反应3~3.5h,将所得物料减压过滤,用乙腈洗涤所得固体物料,干燥,得到对称引达省-1,3,5,7(2h,6h)-四酮。
在本发明中,所述1,3,5-三(4-醛基吡啶基)三嗪、对称引达省-1,3,5,7(2h,6h)-四酮、有机溶剂、酸性催化剂和水混合优选是在保护气氛中,将1,3,5-三(4-醛基吡啶基)三嗪、对称引达省-1,3,5,7(2h,6h)-四酮和有机溶剂搅拌混合5~15min,然后向所得混合物料中加入酸性催化剂的水溶液。
本发明对于提供所述保护气氛的保护气体种类没有特殊的限定,采用本领域技术人员熟知的保护气体即可,具体如氮气。
在本发明中,所述亲核加成-消除反应的温度优选为110~130℃,更优选为120℃;时间优选为70~75h,更优选为72h。
完成所述亲核加成-消除反应后,本发明优选将亲核加成-消除反应完成后所得体系进行固液分离,依次采用n,n-二甲基甲酰胺和乙醇对所得固体物料进行洗涤,干燥,依次采用甲醇和二氯甲烷对所得干燥物料进行索氏提取,干燥,得到具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物。本发明对于所述固液分离的方式没有特殊的限定,采用本领域技术人员熟知的固液分离的技术方案即可,具体如减压抽滤。本发明对于所述洗涤没有特殊的限定,采用本领域技术人员熟知的洗涤的技术方案即可。本发明对于洗涤后进行的干燥没有特殊的限定,采用本领域技术人员熟知的干燥的技术方案、能够将物料充分干燥即可。本发明对于所述索氏提取没有特殊的限定,采用本领域技术人员熟知的索氏提取的技术方案即可;本发明采用甲醇和二氯甲烷对所得干燥物料进行索氏提取,是为了保证充分除去聚合物表面和孔道内部溶剂及未反应的单体。在本发明中,所述索氏提取后进行的干燥优选为真空干燥,所述真空干燥的温度优选为40~60℃,更优选为50℃;时间优选为12~24h,更优选为15~20h。
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
制备1,3,5-三(4-醛基吡啶基)三嗪,包括以下步骤:
将1.1723g三聚氯氰、1.964g4-吡啶醛和80ml四氢呋喃混合,搅拌2h,然后在保护气氛中、70℃条件下反应72h;将所得体系进行减压抽滤,依次采用四氢呋喃和乙醇对所得固体物料进行洗涤,干燥,得到1,3,5-三(4-醛基吡啶基)三嗪。
制备对称引达省-1,3,5,7(2h,6h)-四酮,包括以下步骤:
将4.9810g均苯四甲酸酐、9.0ml乙酰乙酸乙酯和28.0ml三乙胺在搅拌条件下混合,水浴加热至60℃,向所得混合物中加入75.0ml乙酸酐,油浴加热至100℃回流2h;反应结束后冷却至室温,然后在0℃条件下冷却12h,出现淡褐色沉淀物,将所得物料减压抽滤,分别用乙酸酐和乙醚洗涤,得到橙色沉淀物,干燥;将干燥后所得产物按1:100比例加入蒸馏水溶解,在冰水浴条件下,向所得物料中滴加浓硫酸,待溶液变咖啡色,再继续滴加1ml浓硫酸,将所得物料用乙醇进行洗涤,干燥;将干燥后所得产物与120ml乙腈混合,在搅拌、在90℃条件下反应3h,将所得物料减压过滤,用乙腈洗涤所得固体物料,干燥,得到对称引达省-1,3,5,7(2h,6h)-四酮2.3725g,产率48.5%。
制备具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物,包括以下步骤:
在氮气保护下,将0.5138g1,3,5-三(4-醛基吡啶基)三嗪、0.3526g对称引达省-1,3,5,7(2h,6h)-四酮、12ml正丁醇和28ml邻二氯苯混合,然后向所得混合物料中加入4ml乙酸水溶液(由1.4ml乙酸和2.6ml水配制而成),搅拌混合20min,在120℃条件下反应72h,将所得体系进行减压抽滤,依次采用n,n-二甲基甲酰胺和乙醇对所得固体物料进行洗涤,干燥,采用甲醇和二氯甲烷对所得干燥物料进行索氏提取,得到具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物(bhy-cmp)。
对制备得到的1,3,5-三(4-醛基吡啶基)三嗪进行表征,具体如下:
图1为1,3,5-三(4-醛基吡啶基)三嗪的核磁氢谱图,由图1可知,在δ≈7.2处的吸收峰是化合物中1、6、11号位置的质子峰,在δ≈1.0处的吸收峰是化合物2、4、7、9、12、15号位置的质子峰,在δ≈0.1处的吸收峰是化合物3、5、8、10、13、14号位置的质子峰,其余出现的峰属于杂质峰。证明本实施例中所得化合物确实为1,3,5-三(4-醛基吡啶基)三嗪。
图2为1,3,5-三(4-醛基吡啶基)三嗪的红外光谱图,由图2可知,在3116cm-1处的吸收峰符合苯环中c—h伸缩的吸收频率范围,在2783cm-1处的吸收峰符合醛基c—h的吸收频率范围,在1722cm-1处的吸收峰符合醛基中c=o伸缩振动的吸收频率范围,在1593cm-1处的吸收峰符合苯环中的c=c的吸收频率范围,在840cm-1处的吸收峰符合有苯环中c—c的吸收频率范围,在1400cm-1处的吸收峰符合c—n的吸收频率范围。证明本实施例中所得化合物确实为1,3,5-三(4-醛基吡啶基)三嗪。
图3为2,4,6-三(4-甲酰基苯氧基)-1,3,5三嗪的紫外光谱图,由图3可知,化合物的光谱吸收在紫外-可见光范围。
对制备得到的对称引达省-1,3,5,7(2h,6h)-四酮进行表征,具体如下:
图4为对称引达省-1,3,5,7(2h,6h)-四酮的核磁氢谱图,由图4可知,化合物中2、4处的质子峰表现在δ≈7.3处附近,化合物中1、3处的质子峰表现在δ≈1.6处附近,其余出现的峰属于杂质峰。证明本实施例中所得化合物确实为对称引达省-1,3,5,7(2h,6h)-四酮。
图5为对称引达省-1,3,5,7(2h,6h)-四酮的红外光谱图,由图5可知,在1726cm-1处的吸收峰符合c=o的吸收频率范围,在1354cm-1处的吸收峰符合苯环中c=c伸缩的吸收频率范围,在978cm-1处的吸收峰符合苯环中c—c吸收峰的吸收频率范围。证明本实施例中所得化合物确实为对称引达省-1,3,5,7(2h,6h)-四酮。
对制备得到的具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物(bhy-cmp)进行表征,具体如下:
图6为具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的红外光谱图,由图6可知,聚合物在2600.35cm-1处的醛基特征峰消失,在1678.53cm-1处有烯烃的c=c双键吸收峰。证明本实施例中所得化合物确实为具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物。
图7为具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的紫外光谱图,由图7可知,与单体1,3,5-三(4-醛基吡啶基)三嗪相比,目标聚合物的光谱吸收集中在401~872nm范围,表明该单体与对称引达省-1,3,5,7(2h,6h)-四酮聚合后所得聚合物会达到全光谱吸收。
图8为具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的热重分析图,由图8可知,聚合物的失重率为55.3%;具体的,在17~100℃的升温过程中,失重率为15.1%,主要是由于表面吸附水、结构水的脱失而引起的;从100℃开始,样品质量迅速下降,失重率大约为40%,表明聚合物结构在此温度范围内已崩塌并且结构受损。
图9为具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的xrd扫描分析图,由图9可知,本发明提供的共轭微孔聚合物无明显强峰,为无定型结构,属于非晶形材料。
图10为具有式i所示结构的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物的电镜扫描图,由图10可知,本发明提供的共轭微孔聚合物具有片状结构。
由以上实施例可知,本发明提供的基于1,3,5-三(4-醛基吡啶基)三嗪的共轭微孔聚合物呈现片状结构,为非晶形材料,且具备紫外吸收性能,能够应用于光催化等领域。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!