一种基于噻吩并吡咯二酮型小分子受体材料及其制备与应用的制作方法
本发明属于有机半导体材料的技术领域,涉及有机半导体材料的制备方法,特别涉及一种基于噻吩并吡咯二酮a2-π-a1-π-a2结构的小分子受体材料(有机小分子电子传输材料)及其制备方法与应用。
背景技术:
有机半导体材料近年来在有机发光二极管(oleds)、有机薄膜晶体管(ofets)、有机薄膜太阳能电池(opvs)等领域发展迅速,具有重要的应用前景。富勒烯(fullerenes)作为电子受体,广泛地应用于opv器件研究,但其难以修饰。常见富勒烯衍生物pc61bm及pc71bm在可见光区吸收较弱。而非富勒烯受体材料,具有结构多样,可在可见光、近红外区域强吸收等特点,有利于提高opv器件光电转化效率以及稳定性,从而成为opv领域的研发热点。
噻吩并吡咯二酮作为强吸电子单元,可制备具有优良电子传输能力的有机光电材料。当前高效非富勒烯受体材料主要基于idt(茚并二噻吩),端基为苯并茚酮的窄带系受体材料。而基于中等带隙的高效小分子受体材料报道甚少,这会影响活性层材料在400-600nm处的吸收,从而影响器件的光生电流。因此设计制备中等带隙、高迁移率、高性能有机小分子受体材料具有重要的意义。
技术实现要素:
为了克服现有技术的不足,本发明的目的之一在于提供一种噻吩并吡咯二酮型小分子受体材料。本发明的小分子受体材料具有a2-π-a1-π-a2结构。本发明的小分子受体材料迁移率高、合成提纯较简单,同时具有良好的溶解性。
本发明的目的之二在于提供上述基于噻吩并吡咯二酮型小分子受体材料的制备方法。
本发明的再一目的在于提供上述基于噻吩并吡咯二酮型小分子受体材料的应用。所述基于噻吩并吡咯二酮型小分子受体材料在太阳能电池等光电器件中的应用。
本发明的目的通过以下技术方案实现:
基于噻吩并吡咯二酮型小分子受体材料,其结构为a2-π-a1-π-a2,a1为吸电子单元,吸电子单元a1=
其中r为碳数为1~12的烷基链或者烷氧基链中的任意一种;
所述的ar具有以下吸电子单元结构中的任意一种:
其中r1为碳数为1~12的烷基链或者烷氧基链中的任意一种;
吸电子端基a2具有以下任意一种结构:
其中r1为碳数为1~12的烷基链或者烷氧基链中的任意一种;
π桥具有以下任意一种结构:
其中,r2为h、碳数为1~12的烷基链或者烷氧基链中的任意一种。
优选的,所述a2-π-a1-π-a2结构的小分子受体材料为(ehtpdthrcn)2bt,具有以下化学结构:
所述噻吩并吡咯二酮型a2-π-a1-π-a2受体材料的制备方法,包括以下步骤:
(1)制备二溴代中间体br-ar-br:对于ar基团为以下几种结构:
二溴代中间体通过液溴溴代反应制备;
对于ar基团为以下几种结构:
二溴代中间体由nbs溴代反应制备;
(2)在有机溶剂中,将步骤(1)中所得到的二溴代中间体与双联频哪醇硼酸酯在碱和催化剂的作用下发生反应,获得含双硼酸酯的suzuki试剂中间体;所述碱为醋酸钾,所述有机溶剂为无水四氢呋喃或无水1,4-二氧六环,所述催化剂为二(三苯基膦)氯化钯;
(3)步骤(2)中所得到的含双硼酸酯的suzuki试剂中间体和单碘代噻吩并吡咯二酮在催化剂的作用下,生成缺电子的a1中间体单元;所述催化剂为三(二亚苄基丙酮)二钯;
单碘代噻吩并吡咯二酮的结构为:
(4)在有机溶剂中,将步骤(3)中所得到的吸电子的a1中间体单元与n-溴代琥珀酰亚胺在三氟乙酸和浓硫酸条件下反应,得到双溴代中间体br-a1-br;所述有机溶剂为氯仿或四氢呋喃;
(5)步骤(4)中的双溴代中间体br-a1-br与带有缩醛的给电子π桥单元的锡试剂在催化剂作用下,发生stille偶联反应,得到含有缩醛的中间体单元;所述催化剂为三(二亚苄基丙酮)二钯;
带有缩醛的给电子π桥单元的锡试剂:
(6)将步骤(5)中含有缩醛的中间体单元水解,然后与吸电子端基a2单元发生诺文格尔(knoevenagel)反应,得到基于噻吩并吡咯二酮的型a2-π-a1-π-a2小分子受体材料。
优选的,所述的受体材料为(ehtpdthrcn)2bt,具有以下化学结构:
其制备方法包括以下步骤:
(1)4,7-二溴苯并[c][1,2,5]噻二唑的合成:
在氢溴酸溶液中,将苯并[c][1,2,5]噻二唑与液溴在120~130℃下回流反应12~18h,后续处理,获得4,7-二溴苯并[c][1,2,5]噻二唑;
步骤(1)中所述后续处理是指反应完后冷却,在冰浴条件下缓慢加入强碱溶液,调节ph为中性,然后加入二氯甲烷和水萃取,有机层用干燥剂干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为石油醚和二氯甲烷;
其中,苯并[c][1,2,5]噻二唑与液溴的摩尔比为1:(2~2.5);
(2)4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并噻二唑的合成:
在有机溶剂中,将4,7-二溴苯并[c][1,2,5]噻二唑与双联频哪醇硼酸酯在碱和催化剂的作用下反应,后续处理,获得4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并噻二唑;步骤(2)中所述碱为醋酸钾,所述催化剂为二(三苯基膦)二氯化钯;所述有机溶剂为四氢呋喃,所述反应的条件为90~100℃下回流反应12~15h,所述后续处理是指反应完后,冷却后,加入二氯甲烷和水萃取,有机层用干燥剂干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为石油醚和乙酸乙酯;
其中,4,7-二溴苯并[c][1,2,5]噻二唑、醋酸钾、双联频哪醇硼酸酯、二(三苯基膦)二氯化钯的摩尔比为1:(3~4):(2~2.5):(0.03~0.04);
(3)1,1'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)的合成:
在有机溶剂和水中,将4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并噻二唑与5-(2-乙基己基)-1-碘-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮在醋酸钾、三(二亚苄基丙酮)二钯(0)和三(2-甲基苯基)膦的作用下反应,后续处理,获得1,1'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮);步骤(3)中所述反应的条件为100~110℃回流反应12~18h;所述后续处理是指反应完后,冷却,加入二氯甲烷和蒸馏水萃取,有机层用干燥剂干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚;所述有机溶剂为四氢呋喃;
其中,4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并噻二唑、5-(2-乙基己基)-1-碘-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮、醋酸钾、三(二亚苄基丙酮)二钯(0)、三(2-甲基苯基)膦的投料摩尔比为1:(2.1~2.5):(6~10):(0.03~0.07):(0.24~0.54);
(4)3,3'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-溴-5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)的合成:
在有机溶剂中,将1,1'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)与n-溴代琥珀酰亚胺在三氟乙酸和浓硫酸的作用下闭光反应1.5~2h,后续处理,获得3,3'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-溴-5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮);步骤(4)中所述后续处理是指反应完后,缓慢加入氢氧化钠溶液,调节ph为中性,然后加入二氯甲烷和水萃取,有机层用干燥剂干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚;所述有机溶剂为氯仿;
其中,1,1'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)、n-溴代琥珀酰亚胺、三氟乙酸、浓硫酸的投料摩尔比为1:(2.1~3):(130~200):(90~150);
(5)5,5'-(苯并[c][1,2,5]-4,7-二基双(5-(2-乙基己基)-4,6-二酮-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-2-甲醛)的合成:
在有机溶剂中,将3,3'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-溴-5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)与(5-(1,3-二氧戊环2-基)-3-辛基噻吩-2-基)三丁基锡烷在三(二亚苄基丙酮)二钯(0)和三(2-甲基苯基)膦的作用下反应,后续处理,获得产物;然后将此产物进行水解,获得5,5'-(苯并[c][1,2,5]-4,7-二基双(5-(2-乙基己基)-4,6-二酮-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-2-甲醛);步骤(5)中所述有机溶剂为甲苯,所述反应的条件为在110~120℃下回流反应24~36h,所述后续处理是指冷却后,加入氟化钾溶液,继续搅拌,抽滤滤去不溶固体,在液相中加入二氯甲烷萃取,有机层用干燥剂干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚;
所述水解是指在有机溶剂中,产物在盐酸水溶液的作用下,于90~100℃回流12~15h,冷却后,加入二氯甲烷和水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚,得到暗红色固体;所述有机溶剂为四氢呋喃,所述盐酸水溶液是由12mol/l盐酸和去水按体积比为2~3:1混合得到;
其中,3,3'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-溴-5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)、(5-(1,3-二氧戊环2-基)-3-辛基噻吩-2-基)三丁基锡烷、三(二亚苄基丙酮)二钯(0)、三(2-甲基苯基)膦、盐酸的投料摩尔比为1:(2.1~3):(0.03~0.06):(0.24~0.48);(210~2500);
(6)2,2'-((5e,5'e)-(((苯并[c][1,2,5]噻二唑-4,7-二基双(5-(2-乙基己基)-4,6-二氧-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-5,2-二基))双(甲烷取代基))双(3-乙基-4-氧代四氢噻唑-5,2-取代基))二丙二腈的合成:
在有机溶剂中,将5,5'-(苯并[c][1,2,5]-4,7-二基双(5-(2-乙基己基)-4,6-二酮-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-2-甲醛)与2-(3-乙基-4-氧代噻唑-2-亚甲基)丙二腈在醋酸铵和冰乙酸的作用下回流反应,获得受体材料。
步骤(6)中所述回流反应的条件为在85~90℃下回流反应24~36h;回流反应完成后冷却,加入二氯甲烷和水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷;所述有机溶剂为1,2-二氯乙烷;
其中,5,5'-(苯并[c][1,2,5]-4,7-二基双(5-(2-乙基己基)-4,6-二酮-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-2-甲醛)、2-(3-乙基-4-氧代噻唑-2-亚甲基)丙二腈、醋酸铵的投料摩尔比为1:(2.1~2.5):(3~4)。
所述的基于噻吩并吡咯二酮a2-π-a1-π-a2结构的小分子受体材料的应用,用于制备太阳能电池器件。
本发明的材料合成提纯简单,具有良好的溶解性、成膜性、薄膜形貌稳定性以及电子迁移率,并且该小分子受体材料与coi8dfic具有良好的混溶性,在太阳能电池器件中,作为受体,具有重要的应用前景。
与现有技术相比,本发明具有以下优点和有益效果:
(1)本发明引入噻吩并吡咯二酮单元,构建吸电性单元a1,由此设计制备了具有强吸收、中等带隙结构的a2-π-a1-π-a2型小分子受体材料。
(2)本发明制备的基于噻吩并吡咯二酮小分子受体材料具有良好的溶解性、成膜性、薄膜形貌稳定性以及电子迁移率等,有利于获得高效稳定的的溶液加工型有机光伏器件。
(3)本发明制备的基于噻吩并吡咯二酮小分子受体材料不仅能够用作有机太阳能电池受体材料,同时可能作为钙钛矿太阳能电池中的电子传输材料。
(4)本发明制备的基于噻吩并吡咯二酮小分子受体材料合成提纯较简单,可以实现大量制备,在太阳能电池器件中具有重要的应用前景。
附图说明
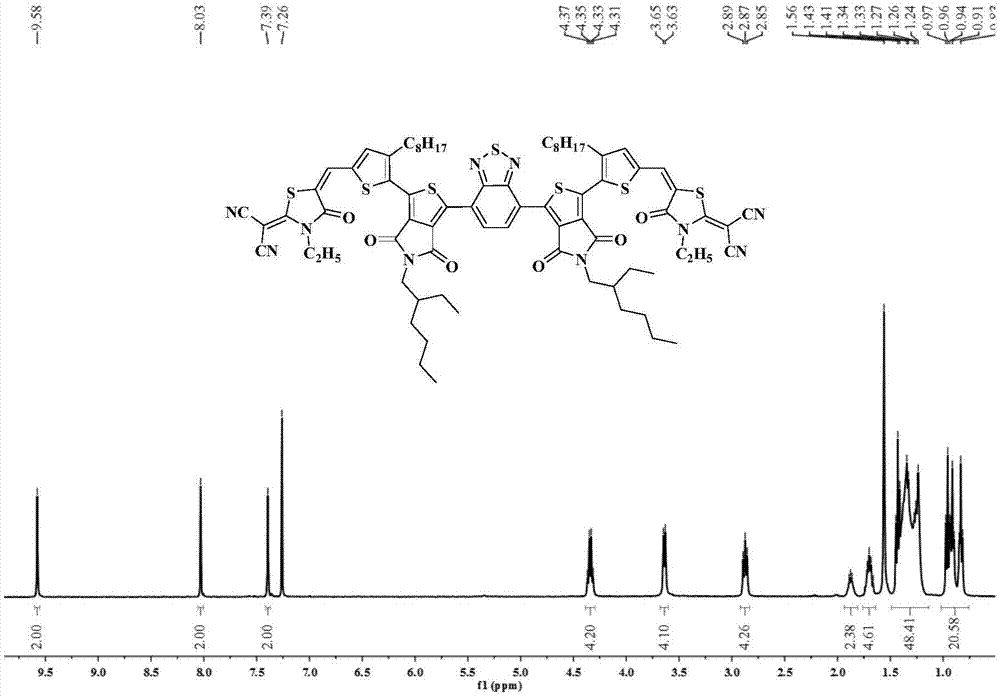
图1为本发明的实施例1制备的基于噻吩并吡咯二酮a2-π-a1-π-a2型结构的小分子受体材料(ehtpdthrcn)2bt的核磁共振氢谱图;
图2为本发明的(ehtpdthrcn)2bt的溶液及薄膜吸收光谱图;
图3为本发明的(ehtpdthrcn)2bt的热失重(tga)曲线;
图4为本发明的(ehtpdthrcn)2bt的差示扫描量热分析(dsc)曲线;
图5为本发明的(ehtpdthrcn)2bt的电化学曲线;
图6为本发明的(ehtpdthrcn)2bt的单电子器件的空间电荷限制电流(sclc)与电压特性曲线(器件结构:ito/zno/(ehtpdthrcn)2bt/pfn-br/al);
图7为(ehtpdthrcn)2bt的有机光伏器件的电流-电压曲线(器件结构为ito/zno/ptb7-th:(ehtpdthrcn)2bt(100nm)/moo3/al);
图8是实施例1制备的(ehtpdthrcn)2bt的有机光伏器件在不同退火温度下的电流-电压(j-v)曲线(器件结构:ito/zno/ptb7-th:(ehtpdthrcn)2bt(100nm)/moo3/al);
图9为本发明的(ehtpdthrcn)2bt的有机光伏器件的外量子效率(eqe)曲线;
图10为本发明的(ehtpdthrcn)2bt的有机光伏器件的有效电压-饱和电流曲线;
图11为本发明的(ehtpdthrcn)2bt的有机光伏器件的光强-电流曲线;
图12为(ehtpdthrcn)2bt的三元有机光伏器件的电流-电压曲线(器件结构为ito/zno/ptb7-th:(ehtpdthrcn)2bt:coi8dfic(100nm)/moo3/al)。
具体实施方式
下面结合实施例,对本发明作进一步地详细说明,但本发明的实施方式不限于此。
实施例1
本实施例的基于噻吩并吡咯二酮(ehtpdthrcn)2bt小分子受体材料材料的结构式:
其合成步骤如下:
步骤一化合物4,7-二溴苯并[c][1,2,5]噻二唑的合成
将苯并[c][1,2,5]噻二唑(10g,0.073mol)加入到150ml氢溴酸溶液(8.7mol/l的氢溴酸溶液)中,用恒压滴液漏斗缓慢加入液溴(25.66g,0.1606mol),加完后,反应在130℃下回流反应12h,冷却后,在冰浴条件下缓慢加入氢氧化钠溶液,调节ph为7左右,然后加入二氯甲烷和蒸馏水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为石油醚和二氯甲烷,得到淡黄色晶体19.7g,产率92%。1hnmr(500mhz,cdcl3,ppm):δ8.13(s,2h)。
步骤二化合物4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并噻二唑的合成
在氮气保护下,将4,7-二溴苯并[c][1,2,5]噻二唑(1g,3.41mmol),双联频哪醇硼酸酯(1.98g,7.8mmol),醋酸钾(1.34g,13.64mmol)加入到40ml的无水四氢呋喃中并搅拌,通入氮气30min后,迅速加入二(三苯基膦)二氯化钯(71.8mg,0.102mmol),反应在90℃下回流反应12h,冷却后,加入二氯甲烷和蒸馏水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为石油醚和乙酸乙酯,得到米黄色固体1g,产率75.7%。
1hnmr(500mhz,cdcl3,ppm):δ8.13(s,2h),1.44(s,24h)。
步骤三化合物1,1'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)合成
将4,7-双(4,4,5,5-四甲基-1,3,2-二氧硼杂环戊烷-2-基)苯并噻二唑(180mg,0.46mmol),5-(2-乙基己基)-1-碘-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮(400mg,1.02mmol),醋酸钾(451mg,4.6mmol)加入到20ml四氢呋喃中并搅拌,然后加入2ml蒸馏水,通入氮气30min后,迅速加入三(二亚苄基丙酮)二钯(0)(28mg,0.0306mmol),三(2-甲基苯基)膦(74mg,0.2448mmol),反应在100℃下回流反应12h,冷却后,加入二氯甲烷和蒸馏水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚,然后用甲醇重结晶纯化,得到橙红色固体210mg,产率69%。
1hnmr(500mhz,cdcl3,ppm):δ9.43(s,2h),7.99(s,2h),3.59(t,j=15.0hz,4h),1.87(s,2h),1.47-1.20(m,16h),1.04-0.81(m,12h)。
步骤四化合物3,3'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-溴-5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)的合成
将1,1'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)(250mg,0.377mmol)加入到30ml氯仿中并搅拌,依次加入三氟乙酸(4ml),浓硫酸(2ml),n-溴代琥珀酰亚胺(201mg,1.131mmol),在闭光条件下反应2h,反应完后,缓慢加入去离子水和氢氧化钠固体,调节ph为7左右,然后加入二氯甲烷和蒸馏水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,随后用甲醇重结晶纯化,得到红色固体300mg,产率96.7%。
1hnmr(500mhz,cdcl3,ppm):δ9.38(s,2h),3.59(t,j=10.5hz,4h),1.87(s,2h),1.47-1.20(m,16h),1.04-0.81(m,12h)
步骤五化合物5,5'-(苯并[c][1,2,5]-4,7-二基双(5-(2-乙基己基)-4,6-二酮-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-2-甲醛)的合成
将3,3'-(苯并[c][1,2,5]噻二唑-4,7-二基)双(1-溴-5-(2-乙基己基)-4h-噻吩[3,4-c]吡咯-4,6(5h)-二酮)(200mg,0.243mmol),(5-(1,3-二氧戊环2-基)-3-辛基噻吩-2-基)三丁基锡烷(407.5mg,0.73mmol),加入到25ml甲苯中并搅拌,通入氮气30min,迅速加入三(二亚苄基丙酮)二钯(0)(13.35mg,0.0146mmol),三(2-甲基苯基)膦(35.5mg,0.1168mmol),反应在110℃下回流反应24h,冷却后,加入氟化钾溶液,继续搅拌20min,抽滤滤去不溶固体,然后在液相中加入二氯甲烷萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚,得到暗红色固体;然后将此产物加入到20ml四氢呋喃中,加入10ml盐酸和5ml去离子水,反应加热到90℃回流12h,冷却后,加入二氯甲烷和蒸馏水萃取,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷和石油醚,然后用甲醇重结晶,得到暗红色固体150mg,产率55.6%。
1hnmr(500mhz,cdcl3,ppm):δ9.93(d,j=5.2hz,2h),9.55(s,2h),7.72(s,2h),3.67-3.56(m,4h),2.89-2.79(m,4h),1.86(dt,j=12.3,6.2hz,2h),1.78-1.65(m,4h),1.47-1.16(m,36h),1.03-0.77(m,18h)。
步骤六化合物(ehtpdthrcn)2bt的合成
将5,5'-(苯并[c][1,2,5]-4,7-二基双(5-(2-乙基己基)-4,6-二酮-5,6-二氢-4h-噻吩[3,4-c]吡咯-3,1-二基))双(4-辛基噻吩-2-甲醛)(150mg,0.135mmol),2-(3-乙基-4-氧代噻唑-2-亚甲基)丙二腈(65.4mg,0.338mmol),加入到30ml1,2-二氯乙烷中并搅拌,通入氮气30min,然后加入醋酸铵(31.21mg,0.405mmol)和冰乙酸(3ml),反应在85℃下回流反应24h,冷却后,加入二氯甲烷和蒸馏水萃取3次,有机层用无水硫酸镁干燥、过滤、减压蒸馏除去溶剂后用硅胶柱分离,洗脱剂为二氯甲烷,经甲醇重结晶后,得到紫黑色固体150mg,产率76.5%。
1hnmr(400mhz,cdcl3,ppm):δ9.58(s,2h),8.03(s,2h),7.39(s,2h),4.34(q,j=7.0hz,4h),3.64(d,j=7.2hz,4h),2.91-2.84(m,4h),1.92-1.83(m,2h),1.76-1.64(m,4h),1.48-1.17(m,36h),1.00-0.79(m,24h).
测试例
以实施例1制备的材料,制作单电子器件ito/zno/(ehtpdthrcn)2bt/pfn-br(10nm)/al和有机光伏器件ito/zno/ptb7-th:(ehtpdthrcn)2bt/moo3/al。
单电子器件制作过程如下:ito导电玻璃依次经丙酮、洗涤剂、去离子水和异丙醇通过超声清洗,每步20min。在烘箱中烘干后,采用plasma氧等离子处理4min。然后在上述处理过的ito玻璃上,旋涂zno(溶胶-凝胶法),厚度30-40nm,转速为3000rpm,200℃退火1h。然后,在zno表面旋涂旋涂一层(ehtpdthrcn)2bt的氯仿溶液(总浓度16mgml-1,转速为2000rpm,时间为30s,厚度为120nm)。再旋涂上约10nm厚的阴极界面材料pfn-br,最后,在<2×10-4pa的真空下,蒸镀金属al。除zno薄膜的制备过程是在大气环境中完成的,其余所有环节均在氮气气氛的手套箱内完成。
有机光伏器件的制备过程:旋涂zno(溶胶-凝胶法)后,在氮气气氛的手套箱中将活性层ptb7-th:(ehtpdthrcn)2bt的氯仿溶液旋涂在zno上(总浓度16mgml-1,转速为3200rpm,时间为30s,厚度为100nm),然后在<2×10-4pa的真空下,蒸镀moo3(蒸镀速率为
图1是实施例1制备的(ehtpdthrcn)2bt的核磁共振氢谱图。
图2是实施例1制备的(ehtpdthrcn)2bt的溶液及薄膜吸收光谱图。材料在固态时候呈现出较强的分子间π-π堆积作用。
图3是实施例1制备的(ehtpdthrcn)2bt的热失重(tga)曲线。材料分解温度大于314℃,具有较好的热稳定性。
图4是实施例1制备的(ehtpdthrcn)2bt的差示扫描量热分析(dsc)曲线。在294℃时材料表现较强的结晶熔融峰。
图5是实施例1制备的(ehtpdthrcn)2bt的电化学曲线,起始还原电位为-0.85v。
图6是实施例1制备的(ehtpdthrcn)2bt的单电子器件的空间电荷限制电流与电压特性曲线(器件结构:ito/zno/(ehtpdthrcn)2bt/pfn-br/al)。采用空间电荷限制电流(sclc)模型,测试其电子迁移率约为1.28×10-3cm2v-1s-1。
图7是实施例1制备的(ehtpdthrcn)2bt的有机光伏器件的电流-电压(j-v)曲线(器件结构:ito/zno/ptb7-th:(ehtpdthrcn)2bt(100nm)/moo3/al),其中给受体比例分别为:1:1.6,1:1.8,1:12。
图8是实施例1制备的(ehtpdthrcn)2bt的有机光伏器件的电流-电压(j-v)曲线(器件结构:ito/zno/ptb7-th:(ehtpdthrcn)2bt(100nm)/moo3/al),分别在80、100、120℃热退火10分钟。
图9是实施例1制备的(ehtpdthrcn)2bt的有机光伏器件的外量子效率(eqe)曲线,表示产生的电子数/入射的光子数,其大小为积分电流。
图10是实施例1制备的(ehtpdthrcn)2bt的有机光伏器件的有效电压-饱和电流曲线。
图11是实施例1制备的(ehtpdthrcn)2bt的有机光伏器件的光强-电流曲线。
图12为(ehtpdthrcn)2bt的三元有机光伏器件的电流-电压曲线(器件结构为ito/zno/ptb7-th:(ehtpdthrcn)2bt:coi8dfic(100nm)/moo3/al)。
有机光伏特性,在大气条件下,将器件置于标准太阳模拟太阳灯(am1.5g,orielmodel91192)下,使用半导体电池(keithley2400)进行j-v测试。
实施例1制备的(ehtpdthrcn)2bt在光伏器件以及光伏三元器件中的光电性能见表1和表2。
实施例1制备的(ehtpdthrcn)2bt小分子受体材料与ptb7-th、coi8dfic及其混合薄膜的表面能参数见表3。
所制备倒装光伏器件(ito/zno/ptb7-th:(ehtpdthrcn)2bt/moo3/al),在未使用高沸点溶剂添加剂、热退火以及溶剂蒸气退火情况下,获得了6.42%的光电转换效率(pce)。活性层在80℃、100℃、120℃退火10min后,pce分别为6.41%、6.21%、6.08%(表1)。所制备倒装光伏三元器件(ito/zno/ptb7-th:(ehtpdthrcn)2bt:coi8dfic/moo3/al),在未使用高沸点溶剂添加剂、热退火以及溶剂蒸气退火情况下,获得了11.52%的光电转换效率(表2)。
表1倒装光伏器件的光电性能参数
a,b,c分别指在80、100、120℃热退火10分钟。
表2倒装光伏三元器件的光电性能参数
表3接触角以及表面能参数
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受所述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!