新型人源γ-氨基丁酸A型受体激动剂及其制备方法与流程
本发明涉及一类γ-氨基丁酸a型受体激动剂及其制备方法,该类受体激动剂可用于对抑郁症、学习和记忆力衰退、癫痫等相关疾病的治疗。
背景技术:
γ-氨基丁酸a型受体(gabaar)是γ-氨基丁酸门控的氯离子通道,也是基底神经节主要的抑制性神经递质受体,与抑郁症、学习和记忆力衰退、焦虑等疾病密切相关。自从1970年curtis等发现内源性的抑制性神经递质γ-氨基丁酸以来,一系列的γ-氨基丁酸a型受体激动剂相继报导出来,在动物实验和临床上均有一定的抗焦虑,镇痛、抑郁等作用。然而随着用药时间的延长,临床上病人逐渐对这些激动剂产生了耐药性,因此急需开发出新型的高活性、高选择性人源γ-氨基丁酸a型受体激动剂来取代现有药物。
技术实现要素:
本发明所要解决的一个技术问题是针对现有技术的现状提供一类γ-氨基丁酸a型受体激动剂,该类受体激动剂可用于对抑郁症、学习和记忆力衰退、癫痫等相关疾病的治疗。
本发明所要解决的另一个技术问题是针对现有技术的现状提供一种制备上述一类γ-氨基丁酸a型受体激动剂的制备方法。
本发明解决上述技术问题所采用的技术方案为:该新型人源γ-氨基丁酸a型受体激动剂,其特征在于:该γ-氨基丁酸a型受体激动剂为6-甲氧基喹诺林溴代铵盐化合物,包含r1、r2和r3三组结构体系:
式中
r1独立地选自链溴代烷烃、溴代烷醇、溴代烯烃或溴代烯醇;
r2独立地选自直链酯基、卤代直链酯基、苯甲酯基、卤代苯甲酯基、磺酸酯基或卤代磺酸酯基;
r3独立地选自甲基、乙基、丙基或苯基。
优选地,所述的受体激动剂为c4:6-甲氧基-1-(3-(硫代甲基磺酰基)丙基)喹诺林溴化铵盐。
优选地,所述的受体激动剂为c5:溴代6-甲氧基-1-丙基喹诺林铵盐。
优选地,所述的受体激动剂为c6:溴代6-甲氧基-1-丁基喹诺林铵盐。
优选地,所述的受体激动剂为c7:溴代6-甲氧基-1-(2-乙酸基)丙基喹诺林铵盐。
优选地,所述的受体激动剂为c8:6-甲氧基-1-(3-(甲基磺酰氧基)丙基)喹诺林溴化铵盐。
优选地,所述的受体激动剂为c9:溴代6-甲氧基-1-(2-(4-氯苯基)磺酸基)丙基喹诺林铵盐。
本发明为解决第二个技术问题而提供一种上述的c4新型人源γ-氨基丁酸a型受体激动剂的制备方法,其特征在于包括以下步骤:
(1)将5-10g6-甲基喹诺林和7.86-15.72g3-溴-1-丙醇加入到烧瓶中,用100-200ml甲苯溶解,100℃油浴下磁力搅拌反应12h,随着反应的进行,瓶底析出白色固体后,弃去反应液,用10-20ml甲苯洗两次,减压浓缩,得到7.57-15.14g白色固体6-甲氧基-1-(3-羟基丙基)喹诺林铵盐c2;
(2)取2~4g步骤(1)的产物c2置于烧瓶中,加入50-100mlch2cl2,磁力搅拌溶解,氮气保护下用冰盐浴降温至-10~-5℃,再缓慢滴加5.45-10.9gpbr3的ch2cl2溶液,保温反应1h后,室温反应3h,反应液倒入含100ml冰水的烧杯中,玻璃棒搅拌,待无白烟冒出,将水层和油层均倒入1l分液漏斗中,用30ml的ch2cl2萃取三次,有机层用15ml的饱和食盐水洗涤三次,无水硫酸钠干燥,减压浓缩,经由乙酸乙酯和甲醇构成的硅胶柱纯化,乙酸乙酯和甲醇的体积比为1:10,真空干燥12h,得0.86-1.72g棕黄色油状液体溴代6-甲氧基-1-(3-溴丙基)喹诺林铵盐c3;
(3)取0.86-1.72g步骤(2)的棕黄色油状液体c3和0.48-0.96g甲基硫代磺酸钠置于由乙腈和水构成的混合溶剂中,该混合溶剂的乙腈:水的体积比为2:1,室温下磁力搅拌3h,反应液用30ml的ch2cl2萃取三次,采用15ml的饱和食盐水洗涤三次,无水硫酸钠干燥,减压浓缩,经由乙酸乙酯和甲醇构成的硅胶柱纯化,乙酸乙酯和甲醇的体积比为1:20,真空干燥12h,得到0.28-0.56g黄色固体溴代6-甲氧基-1-(2-甲基硫代磺酸基)丙基喹诺林铵盐c4。
本发明为解决第二个技术问题而提供一种上述的c5或c6新型人源γ-氨基丁酸a型受体激动剂的制备方法,其特征在于包括以下步骤:
将6-甲氧基喹诺林(0.5-1g,3.14-6.28mmol)和3-溴丙烷或3-溴丁烷(62.9-125.8mmol)加入到烧瓶中,用30ml甲苯溶解,60℃油浴下磁力搅拌反应12h,反应液用水30ml萃取三次,合并后水相用30ml的ch2cl2反萃取三次,有机层用15ml的饱和食盐水洗涤三次,无水硫酸钠干燥,减压浓缩,经由乙酸乙酯和甲醇构成的硅胶柱纯化,乙酸乙酯和甲醇的体积比为1:10,真空干燥12h,得棕黄色油状液体c5或c6。
本发明为解决第二个技术问题而提供一种上述的c7或c8或c9新型人源γ-氨基丁酸a型受体激动剂的制备方法,其特征在于包括以下步骤:
(1)将5-10g6-甲基喹诺林和7.86-15.72g3-溴-1-丙醇加入到烧瓶中,用100-200ml甲苯溶解,100℃油浴下磁力搅拌反应12h,随着反应的进行,瓶底析出白色固体后,弃去反应液,用10-20ml甲苯洗两次,减压浓缩,得到7.57-15.14g白色固体6-甲氧基-1-(3-羟基丙基)喹诺林铵盐c2;
(2)取0.5-1g步骤(1)的产物c2和0.34-0.68g三乙胺置于烧瓶中,加入20-40mlch2cl2,磁力搅拌溶解,冰浴降温至0℃,再缓慢滴加0.2-0.4g乙酰氯的ch2cl2溶液,保温反应1h,后室温反应2h,反应液倒入含100ml冰水的烧杯中,玻璃棒搅拌,待无白烟冒出,将水层和油层均倒入250ml分液漏斗中,用30ml的ch2cl2萃取三次,采用15ml的饱和食盐水洗涤三次,无水硫酸钠干燥,减压浓缩,经由乙酸乙酯和甲醇构成的硅胶柱纯化,乙酸乙酯和甲醇的体积比为1:10,真空干燥12h,得0.28-0.56g棕黄色油状液体c7或c8或c9。
与现有技术相比,本发明具有如下优点:基于γ-氨基丁酸自身结构特点,结合分子结构设计和同源建模进行初步结构筛选,本发明以6-甲氧基喹诺林为起始原料,在氨基端引入直链烷醇、直链酯基、直链芳香基等基团,合成一系列不同结构特征的激动剂,应用电压钳技术进行体外活性初步筛选,得到2个高活性的激动剂c4和c8,可作为潜在的人源γ-氨基丁酸a型受体激动剂应用于临床。
附图说明
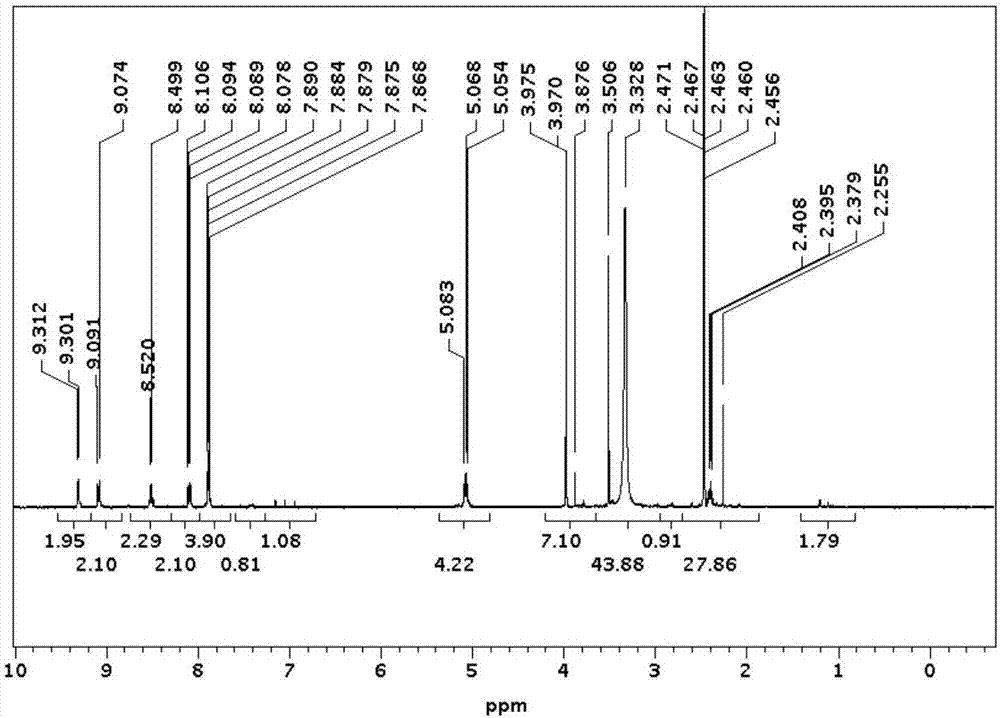
图1为本发明实施例1中化合物c4的氢谱图;
图2为本发明实施例1中化合物c4的质谱图;
图3为本发明实施例1中化合物c4激活α1β2γ2γ-氨基丁酸受体的浓度效应关系图;
图4为本发明实施例1中化合物c4激活α1β2γ2γ-氨基丁酸受体的剂量效应曲线图;
图5为本发明实施例1中竞争性抑制剂荷包牡丹碱对化合物c4的竞争性抑制浓度效应关系图;
图6为本发明实施例1中竞争性抑制剂荷包牡丹碱对化合物c4的竞争性抑制剂量效应曲线图。
具体实施方式
以下通过结合附图实施例对本发明作进一步说明。
实施例16-甲氧基喹诺林溴代铵盐化合物c4的合成
a、化合物c4的合成总体的反应路线以及实验条件如下所示:
b、该化合物溴代6-甲氧基-1-(2-甲基硫代磺酸基)丙基喹诺林铵盐(c4)的制备方法如下:
将5g、31.45mmol的6-甲基喹诺林和7.86、62.9mmol的3-溴-1-丙醇加入到250ml圆底烧瓶中,用100ml甲苯溶解,100℃油浴下磁力搅拌反应12h。随着反应的进行,瓶底析出白色固体。反应结束,弃去反应液,用10ml的甲苯洗两次,减压浓缩,得到7.57g白色固体c2;
c、该白色固体6-甲氧基-1-(3-羟基丙基)喹诺林铵盐c2的1h-nmr谱图:
(c2):81%yield;whitesolid;1h-nmr(500mhz,cdcl3):δ9.38(d,1h,j=5.4hz,ar-h),9.10(d,1h,j=8.9hz,ar-h),8.47(d,1h,j=10.0hz,ar-h),8.12(m,1h,ar-h),7.931(m,2h,ar-h),5.08(m,2h,n-ch2),4.01(s,3h,ch3o),3.51(m,1h,oh),3.36(s,3h,ch2oh),2.11(m,2h,ch2ch2ch2);msmaldim/z:297.04(m)+(100%);
d、将上述产物c2取2g、6.73mmol置于250ml圆底烧瓶中,加入50mlch2cl2,磁力搅拌溶解,氮气保护下用冰盐浴降温至-10~-5
e、将棕黄色油状液体c3(0.86g,2.39mmol)和甲基硫代磺酸钠(0.48g,3.59mmol)置于乙腈和水(v乙腈:v水=2:1)的混合溶剂中,室温下磁力搅拌3h。反应液用ch2cl2(3×30ml)萃取,饱和食盐水(3×15ml)洗涤,无水硫酸钠干燥,减压浓缩,经硅胶柱(v乙酸乙酯:v甲醇=1:20)纯化,真空干燥12h,得到0.28g黄色固体c4。
溴代6-甲氧基-1-(2-甲基硫代磺酸基)丙基喹诺林铵盐(c4)的质谱图:30%yield;yellowsolid;1h-nmr(500mhz,cdcl3):δ9.3(d,1h,j=5.5hz,ar-h),9.1(d,1h,j=8.5hz,ar-h),8.51(d,1h,j=10.5hz,ar-h),8.09(m,1h,ar-h),7.88(m,2h,ar-h),5.07(m,2h,n-ch2),3.97(s,3h,ch3o),3.33(s,3h,ch3s=o),2.39(m,2h,ch2s),2.25(m,2h,ch2ch2);msmaldim/z:394.02(m)+(100%)。
实施例2溴代6-甲氧基-1-(2-甲基硫代磺酸基)丙基喹诺林铵盐(c4)的制备方法如下:
a、将10g、62.90mmol的6-甲基喹诺林和15.72、125.8mmol的3-溴-1-丙醇加入到250ml圆底烧瓶中,用100ml甲苯溶解,100℃油浴下磁力搅拌反应12h。随着反应的进行,瓶底析出白色固体。反应结束,弃去反应液,用10ml的甲苯洗两次,减压浓缩,得到15.14g白色固体c2;
b、将上述产物c2取4g、13.46mmol置于250ml圆底烧瓶中,加入50mlch2cl2,磁力搅拌溶解,氮气保护下用冰盐浴降温至-10~-5
c、将棕黄色油状液体c3(1.72g,4.78mmol)和甲基硫代磺酸钠(0.96g,7.18mmol)置于乙腈和水(v乙腈:v水=2:1)的混合溶剂中,室温下磁力搅拌3h。反应液用ch2cl2(3×30ml)萃取,饱和食盐水(3×15ml)洗涤,无水硫酸钠干燥,减压浓缩,经硅胶柱(v乙酸乙酯:v甲醇=1:20)纯化,真空干燥12h,得到0.56g黄色固体c4。
实施例3溴代6-甲氧基-1-(2-甲基硫代磺酸基)丙基喹诺林铵盐(c4)的制备方法如下:
a、将7g、44.9mmol的6-甲基喹诺林和11.22、89.85mmol的3-溴-1-丙醇加入到250ml圆底烧瓶中,用100ml甲苯溶解,100℃油浴下磁力搅拌反应12h。随着反应的进行,瓶底析出白色固体。反应结束,弃去反应液,用10ml的甲苯洗两次,减压浓缩,得到10.81g白色固体c2;
b、将上述产物c2取2.85g、9.61mmol置于250ml圆底烧瓶中,加入50mlch2cl2,磁力搅拌溶解,氮气保护下用冰盐浴降温至-10~-5
c、将棕黄色油状液体c3(1.22g,3.41mmol)和甲基硫代磺酸钠(0.68g,5.12mmol)置于乙腈和水(v乙腈:v水=2:1)的混合溶剂中,室温下磁力搅拌3h。反应液用ch2cl2(3×30ml)萃取,饱和食盐水(3×15ml)洗涤,无水硫酸钠干燥,减压浓缩,经硅胶柱(v乙酸乙酯:v甲醇=1:20)纯化,真空干燥12h,得到0.4g黄色固体c4。
实施例4化合物溴代6-甲氧基-1-丙基喹诺林铵盐(c5)
将6-甲氧基喹诺林(0.5g,3.14mmol)和3-溴丙烷(0.77g,62.9mmol)加入到100ml圆底烧瓶中,用30ml甲苯溶解,60℃油浴下磁力搅拌反应12h,反应液用水(3×30ml)萃取,合并后水相用ch2cl2(3×30ml)反萃取,有机层用饱和食盐水(3×15ml)洗涤,无水硫酸钠干燥,减压浓缩,经硅胶柱(v乙酸乙酯:v甲醇=1:10)纯化,真空干燥12h,得0.58g棕黄色油状液体c5。
溴代6-甲氧基-1-丙基喹诺林铵盐(c5):65.5%yield;yellowliquid;1h-nmr(500mhz,cdcl3):δ9.35(d,1h,j=5.0hz,ar-h),9.16(d,1h,j=8.0hz,ar-h),8.70(d,1h,j=10.5hz,ar-h),8.15(m,1h,ar-h),7.87(m,2h,ar-h),4.93(m,2h,n-ch2),4.02(s,3h,ch3o),1.91(m,2h,ch2ch3),0.9(s,3h,ch2ch3);msmaldim/z:281.04(m)+(100%)。
实施例5化合物溴代6-甲氧基-1-丙基喹诺林铵盐(c5)
将6-甲氧基喹诺林(1g,6.28mmol)和3-溴丙烷(1.54g,125.8mmol)加入到100ml圆底烧瓶中,用30ml甲苯溶解,60℃油浴下磁力搅拌反应12h,反应液用水(3×30ml)萃取,合并后水相用ch2cl2(3×30ml)反萃取,有机层用饱和食盐水(3×15ml)洗涤,无水硫酸钠干燥,减压浓缩,经硅胶柱(v乙酸乙酯:v甲醇=1:10)纯化,真空干燥12h,得1.16g棕黄色油状液体c5。
实施例6化合物溴代6-甲氧基-1-丙基喹诺林铵盐(c5)
将6-甲氧基喹诺林(0.66g,4.14mmol)和3-溴丙烷(1.01g,83.02mmol)加入到100ml圆底烧瓶中,用30ml甲苯溶解,60℃油浴下磁力搅拌反应12h,反应液用水(3×30ml)萃取,合并后水相用ch2cl2(3×30ml)反萃取,有机层用饱和食盐水(3×15ml)洗涤,无水硫酸钠干燥,减压浓缩,经硅胶柱(v乙酸乙酯:v甲醇=1:10)纯化,真空干燥12h,得0.76g棕黄色油状液体c5。
实施例7化合物溴代6-甲氧基-1-丁基喹诺林铵盐(c6)的制备
化合物c6的制备方法同实施例6的c5相同,经硅胶柱(v乙酸乙酯:v甲醇=1:10)纯化,真空干燥12h,得0.47g棕黄色油状液体c6。
溴代6-甲氧基-1-丁基喹诺林铵盐(c6):50.7%yield;yellowliquid;1h-nmr(500mhz,cdcl3):δ9.38(d,1h,j=5.5hz,ar-h),9.16(d,1h,j=8.7hz,ar-h),8.53(d,1h,j=10.5hz,ar-h),8.15(m,1h,ar-h),7.87(m,2h,ar-h),5.02(m,2h,n-ch2),3.98(s,3h,ch3o),2.34(m,2h,ch2ch3),1.99(m,2h,ch2ch2),0.95(s,3h,ch2ch3);msmaldim/z:295.06(m)+(100%)。
实施例8化合物溴代6-甲氧基-1-(2-乙酸基)丙基喹诺林铵盐(c7)的制备
a、将5g6-甲基喹诺林和7.86g3-溴-1-丙醇加入到烧瓶中,用100ml甲苯溶解,100℃油浴下磁力搅拌反应12h,随着反应的进行,瓶底析出白色固体后,弃去反应液,用10ml甲苯洗两次,减压浓缩,得到7.57g白色固体6-甲氧基-1-(3-羟基丙基)喹诺林铵盐c2;
b、将上述产物c2(0.5g,1.68mmol)和三乙胺(0.34,3.36mmol)置于250ml圆底烧瓶中,加入20mlch2cl2,磁力搅拌溶解,冰浴降温至0
溴代6-甲氧基-1-(2-乙酸基)丙基喹诺林铵盐(c7):48.9%yield;yellowliquid;1h-nmr(500mhz,cdcl3):δ9.39(d,1h,j=5.0hz,ar-h),9.13(d,1h,j=8.0hz,ar-h),8.44(d,1h,j=10.6hz,ar-h),8.13(m,1h,ar-h),7.91(m,2h,ar-h),5.11(m,2h,n-ch2),4.01(s,3h,ch3o),2.42(s,3h,ch3c=o),2.3(m,2h,ch2o),2.01(m,2h,ch2ch2ch2);msmaldim/z:339.05(m)+(100%)。
实施例9化合物溴代6-甲氧基-1-(2-乙酸基)丙基喹诺林铵盐(c7)的制备
a、将10g6-甲基喹诺林和15.72g3-溴-1-丙醇加入到烧瓶中,用100ml甲苯溶解,100℃油浴下磁力搅拌反应12h,随着反应的进行,瓶底析出白色固体后,弃去反应液,用10ml甲苯洗两次,减压浓缩,得到15.14g白色固体6-甲氧基-1-(3-羟基丙基)喹诺林铵盐c2;
b、将上述产物c2(1g,3.36mmol)和三乙胺(0.68,6.72mmol)置于250ml圆底烧瓶中,加入40mlch2cl2,磁力搅拌溶解,冰浴降温至0
实施例10化合物溴代6-甲氧基-1-(2-乙酸基)丙基喹诺林铵盐(c7)的制备
a、将8.3g6-甲基喹诺林和13.04g3-溴-1-丙醇加入到烧瓶中,用100ml甲苯溶解,100℃油浴下磁力搅拌反应12h,随着反应的进行,瓶底析出白色固体后,弃去反应液,用10ml甲苯洗两次,减压浓缩,得到12.56g白色固体6-甲氧基-1-(3-羟基丙基)喹诺林铵盐c2;
b、将上述产物c2(0.83g,2.78mmol)和三乙胺(0.56,5.58mmol)置于250ml圆底烧瓶中,加入20mlch2cl2,磁力搅拌溶解,冰浴降温至0
实施例11化合物溴代6-甲氧基-1-(2-甲基磺酸基)丙基喹诺林铵盐(c8)的制备
其制备方法同实施例10的(c7),该化合物c8分别对c2的羟基进行酯化,在三乙胺存在下与甲磺酰氯和对氯苯磺酰氯反应,经硅胶柱纯化,真空干燥12h,得到白色固体c80.19g。
溴代6-甲氧基-1-(2-甲基磺酸基)丙基喹诺林铵盐(c8):13%yield;palesolid;1h-nmr(500mhz,cdcl3):δ9.38(d,1h,j=5.7hz,ar-h),9.15(d,1h,j=8.6hz,ar-h),8.47(d,1h,j=10.0hz,ar-h),8.13(m,1h,ar-h),7.91(m,2h,ar-h),5.14(m,2h,n-ch2),4.01(s,3h,ch3o),3.36(s,3h,ch3s=o),2.35(m,2h,ch2o),1.90(m,2h,ch2ch2ch2);msmaldim/z:377.21(m)+(100%)。
实施例12化合物溴代6-甲氧基-1-(2-(4-氯苯基)磺酸基)丙基喹诺林铵盐(c9)的制备
制备方法同实施例10的(c7),该化合物c8分别对c2的羟基进行酯化,在三乙胺存在下与甲磺酰氯和对氯苯磺酰氯反应,经硅胶柱纯化,真空干燥12h,得到黄色固体c90.15g。
溴代6-甲氧基-1-(2-(4-氯苯基)磺酸基)丙基喹诺林铵盐(c9):20%yield;yellowsolid;1h-nmr(500mhz,cdcl3):δ9.57(d,1h,j=5.7hz,ar-h),9.23(d,1h,j=8.6hz,ar-h),8.61(d,1h,j=10.0hz,ar-h),8.32(m,1h,ar-h),7.91(m,6h,ar-h),5.44(m,2h,n-ch2),4.45(s,3h,ch3o),2.78(m,2h,ch2o),2.51(m,2h,ch2ch2ch2);msmaldim/z:457.08(m)+(100%)。
实施例13新合成化合物c4-c9的活性初筛
(1)人源γ-氨基丁酸a型受体crna的制备
将人源γ-氨基丁酸a型受体α1、β2、γ2亚基的cdna用phusiondna聚合酶按照快速克隆技术克隆到pgemhe载体上,用nhei内切酶来进行线性化,纯化后用于crna的合成。所述的crna利用t7rna聚合酶进行标准的体外转录,用dnasei(不含rnase)降解dna模板之后,crna经过纯化,保存在焦碳酸二乙酯(depc)处理过的蒸馏水中。crna的产量和完整性用eppendorfbiophotometer和1%的琼脂糖凝胶进行检测。
(2)卵母细胞的制备和注射
卵母细胞从雌性非洲爪蟾(xenopusi,annarbor,mi,usa)中获得,用0.2%ms-222麻醉爪蟾,卵巢叶手术取出并置于孵育液中,孵育液的配方为(mm):82.5nacl,2.5kcl,1mgcl2,1cacl2,1na2hpo4,0.6theophylline,2.5sodiumpyruvate,5hepes,50u/mlpenicillin和50μg/mlstreptomycin,ph7.5。手术后爪蟾腹腔注射止痛剂盐酸塞拉嗪(10mg/kg)和抗生素庆大霉素(3mg/kg)。在放回培养箱之前让它从手术中复苏2-3小时。在第三次手术后,爪蟾将在ms-222麻醉下实行安乐死。手术得到的卵巢叶切成小片,用1wunschunit/ml的liberaseblendzymetm消化,在室温下持续搅拌1.5-2小时。分散的卵母细胞彻底用孵育液漂洗。选择阶段vi的卵母细胞,注射之前在16
(3)双极电压钳
注射crna后2-4天,表达了γ-氨基丁酸a型受体的卵母细胞置于定制小室,持续灌流卵母细胞ring’s液体(or2),其配方为(mm):92.5nacl,2.5kcl,1cacl2,1mgcl2,和5hepes(4-羟乙基哌嗪乙磺酸),ph7.5。该室通过琼脂氯化钾桥接地,以防止任何配体诱导的电位发生变化。卵母细胞钳制在-70mv,用一个axoclamp900a放大器(moleculardevices,sunnyvale,ca,usa)检测γ-氨基丁酸诱导的电流。电流信号在50hz用axoclamp900a内置的4极低通bessel滤波器进行过滤,并用digidata1440a(moleculardevices)和pclamp10在100hz进行数字化。
实施例14新合成化合物c4-c9对α1β2γ2γ-氨基丁酸受体的体外活性
(1)将合成好的化合物c4溶解在甲醇溶液中,然后分装在1.5mleppendorf离心管中,真空干燥10min,除去甲醇溶剂,每管8mg,测活性之前用4
- 还没有人留言评论。精彩留言会获得点赞!