CrGlut1表达基因、表达载体、莱茵衣藻工程藻及制备方法和应用与流程
本发明涉及基因工程技术领域,尤其涉及一种crglut1表达基因、表达载体、莱茵衣藻工程藻及制备方法和应用。
背景技术:
微藻作为生态系统中有机物和能量的最初级提供者,具有环境适应能力强、生长周期短、占地面积小,能合成许多结构和生理功能独特的生物活性物质,和环境效益显著等优点蕴含着巨大的开发价值和经济潜力。在这种背景下,微藻生物技术的研究受到各国政府高度重视,并且对微藻资源的开发利用提出了新的需求,给微藻产业的发展带来了新机遇与挑战。
利用工业发酵技术进行对微藻进行异养化高细胞密度培养是目前微藻工业化生产的一种发展趋势,异养化培养发酵具有以下优点:1)可控制培养条件使微藻生长维持在最佳状态;2)以有机碳源作为培养基成分,大幅度降低了培养成本,提高了生长速度,实现了藻细胞高密度培养;3)采用密闭发酵罐,能够实现无菌培养、单位体积产率高、产品质量可控稳定且藻细胞收集方便等优势。
一些藻类能利用有机物作为唯一碳源和能源进行异养生长,异养化培养微藻,即通过在发酵罐黑暗需氧发酵生长条件下添加葡萄糖等有机碳源对微藻进行培养,达到快速生长且高生物量的目的。近年来对于微藻异养培养的研究已经受到了众多的重视,在对微藻异养方面的研究主要针对于工艺上的优化以达到更快的生长速度和更高的生物量,其中我国研究人员钱峰慧对蛋白核小球藻异养培养基和补料策略进行了优化,在50l发酵罐中通过间歇补料分批培养方式培养100h后,藻细胞密度最高可150g/l。
微藻作为一种光合生物,并非所有的微藻都能进行异养生长,绝大部分生产高附加值产物的微藻在光合生长时的生物量并不能满足大规模工业的需求,因此人们期待通过基因工程技术对微藻进行改造,从而达到使的微藻能够大规模利用有机碳源进行高密度异养生长。
目前关于微藻的异养生长机理的研究还不够深入,其中作为能量来源的糖类物质必须穿过细胞膜进入藻细胞内部,才能被微藻利用,有理论认为由于某些微藻缺乏转运有机物的运输机制,导致微藻只能进行光合自养。zasjavskaia等通过对三角褐指藻(utcc646)进行转运基因的转入,使其带有运输葡萄糖的能力,所用的转运基因包括来自人类红细胞的葡萄糖转运蛋白基因(glut1),来自小球藻的hup1,以及来自酿酒酵母的hxt1、hxt2和hxt4,置于黑暗条件下进行培养,结果显示转入glut1基因的三角褐指藻(utcc646)能够以葡萄糖为唯一碳源并且能够在黑暗条件下生长。
随着基因工程技术的持续发展,通过对有改造潜力的藻类进行定向基因工程改造是一种行之有效的方法。微藻作为生物反应器具有广阔的发展前景和巨大潜力,能够表达具有高活性的蛋白,利用其作为转基因生物反应器通过在可控条件下利用发酵方式生产高附加值产物。因此,以此为基础,构建能够表达出具实质特异性活性蛋白的基因工程藻,非常符合产业和效益的发展需求。
技术实现要素:
本发明的主要目的在于提供一种能够以莱茵衣藻为宿主进行大量表达生产glut1蛋白的crglut1表达基因,旨在利用莱茵衣藻作为转基因生物反应器通过在可控条件下利用发酵方式生产具有生物活性的glut1蛋白产物。
为实现上述目的,本发明提供的crglut1表达基因,具有序列表seq.id.no.1的核苷酸序列。
本发明以上通过对莱茵衣藻和人源密码子使用情况以及glut1基因的密码子偏好性进行分析,并根据莱茵衣藻表达系统密码子偏好性对glut1基因进行优化改造后获得的crglut1表达基因,具有高效表达人源葡萄糖转运蛋白的能力,并且表达生产的glut1蛋白具有生物活性。
本发明进一步还提出由以上crglut1表达基因与载体质粒重组后的重组表达载体、以及转化有重组表达载体的转基因莱茵衣藻。本发明的转基因莱茵衣藻,能够转运胞外葡萄糖,于黑暗条件下能利用葡萄糖生长,并且能大量表达具有生物活性的目的蛋白glut1,具有显著的应用价值。
本发明还提出以上转基因莱茵衣藻的制备方法,包括:以所述的crglut1表达基因为模板进行扩增,获得扩增产物;
将所述扩增产物与phr13质粒分别用限制性内切酶双酶切后,再用连接酶连接,获得重组表达载体;
将所述重组表达载体导入莱茵衣藻工程藻宿主中,即获得转基因莱茵衣藻。
本发明进一步还提出将以上能成功表达crglut1基因的转基因藻在制备保健食品、异养生物反应器、医药、水产养殖饵料或者动物饲料添加剂中的应用。其表达的具有良好生物活性的目的蛋白glut1,作为医疗、免疫、活性添加成分于饵料/饲料、都具有非常良好的应用前景和价值。
附图说明
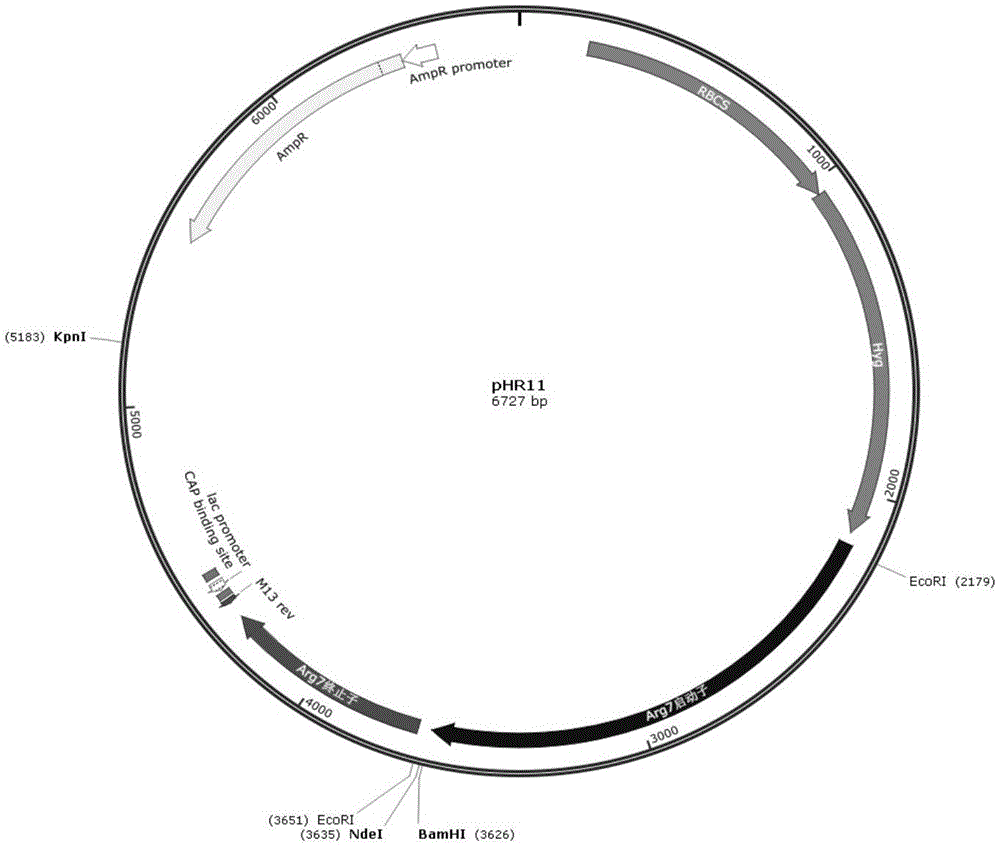
图1为本发明所采用的phr13载体质粒的质粒图谱;
图2为本发明构建的重组表达载体phr13-crglut1的质粒图谱;
图3为本发明实施例重组质粒puc19-crglut1的pcr扩增检测结果图;
图4为本发明实施例重组质粒phr13-crglut1菌落pcr扩增检测结果图;
图5为本发明实施例重组质粒phr13-crglut1的酶切结果验证图;
图6为本发明实施例转crglut1基因衣藻基因组的pcr扩增检测结果图;
图7为本发明实施例转crglut1基因衣藻qrt-pcr检测结果图;
图8为本发明实施例转crglut1基因衣藻western杂交分析结果图;
图9为本发明实施例转crglut1基因衣藻2-nbdg转运检测结果图
图10为本发明实施例转crglut1基因衣藻黑暗异养条件下生物量的检测结果图;
图11为本发明实施例转crglut1基因衣藻兼养条件下生物量的检测结果图。
具体实施方式
本发明目的的实现、功能特点及优点将结合实施例,参照附图做进一步说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明提供一种crglut1表达基因,其主要是用于编码glut1蛋白,后续能作为目的基因片段而通过基因工程手段构建表达载体和工程藻,从而大量表达具有生物活性的目的glut1蛋白。其中,
glut1蛋白是在人体内分布最为广泛的转运体,主要参与人体内葡萄糖的跨膜运输,负责组织与血液之间的葡萄糖运输,作为已知分布最广的转运蛋白,几乎存在于人体每一个细胞膜上,对于维持基础状态下组织细胞的葡萄糖摄入起着关键作用,在医疗(如糖尿病治疗)、免疫等方面具有重要的价值。
基于以上,本发明的crglut1表达基因具有序列表seq.id.no.1的核苷酸序列,核苷酸序列长度为1479bp。本发明提出的crglut1表达基因,通过分析现有的glut1基因,对照莱茵衣藻偏爱密码子表,对莱茵衣藻和人源密码子使用情况以及glut1基因的密码子偏好性进行分析,并根据莱茵衣藻表达系统密码子偏好性对glut1基因进行优化改造后获得,因此命名为crglut1表达基因。
而以上,基于莱茵衣藻存在的表达优势进行,能提升最终表达产物的生物活性。具体,莱茵衣藻是少数拥有可遗传转化的三套基因组,即细胞核,叶绿体和线粒体三套基因组均能进行遗传转化的生物;并且莱茵衣藻核表达的重组蛋白能够进行翻译后加工和修饰如糖基化等,使得莱茵衣藻核表达的重组蛋白具有较高的生物活性;同时莱茵衣藻光合效率高且生长周期短,其细胞分裂周期仅为8~12h,3~4天即可达到对数生长期,且具有外泌蛋白的能力使得后续分离纯化难度大大降低,经济效益明显,且培养过程中不存在类似于植物生物反应器的基因漂流现象,生物安全性能良好。
本发明以上改造获得的crglut1表达基因,具有高效表达人源葡萄糖转运蛋白的能力,并且表达生产的glut1蛋白具有生物活性;同时转化对象藻细胞能够转运胞外葡萄糖,于黑暗条件下能利用葡萄糖生长,具有显著的应用价值。
本发明进一步还提出一种含有以上crglut1表达基因的重组表达载体,通过将以上crglut1表达基因于载体质粒通过基因工程手段重组获得。
实施中,优选重组表达载体包括phr13载体质粒和crglut1表达基因;其中,phr13载体质粒是比较合适于莱茵衣藻中表达的质粒,具体质粒结构图谱可以参见图1所示,含有arg7组成型启动子序列和arg7终止子序列,中间携带有ndei、bamhi、ecori、等多克隆位点,可以插入外源目的基因在莱茵衣藻中持续高效表达,同时载体phr13含有遗传表达盒rbcs2∷hyg∷rbcs2,使转化的藻细胞获得潮霉素抗性,可作为莱茵衣藻遗传转化的筛选标记。
因此,本发明实施中采用phr13载体质粒作为crglut1表达基因的表达载体,而重组之后的表达载体中,crglut1表达基因被配置为受莱茵衣藻组成型启动arg7的调控,从而高效表达。通过基因重组手段获得的重组表达载体,可以名为表达载体phr13-crglut1,其结构可以参见图2所示。
进一步,本发明还提出一种含有以上重组表达载体的基因工程藻,可以通过对基因工程藻大量培养、并诱导crglut1表达基因表达生产glut1蛋白。这一基因工程藻基于本发明以上描述,采用的是以莱茵衣藻为宿主,然后将以上表达载体phr13-crglut1进行转化获得。转化的方法,可以采用基因工程中常用的玻璃珠转化法、电转化法、基因枪法或农杆菌转化法等进行
本发明还提出以上述crglut1表达基因为基础,制备上述莱茵衣藻工程藻的方法,方法步骤包括:
s10,以上述crglut1表达基因为模板进行pcr扩增;
s20,将步骤s10的扩增产物与骨架载体phr13分别用限制性内切酶ndei和bamhi双酶切后,再用连接酶连接,获得重组表达载体phr13-crglut1;
s30,将重组表达载体phr13-crglut1导入莱茵衣藻的受体藻株中,即获得转基因莱茵衣藻;
同时,为了保证能获得成功转化的转基因莱茵衣藻,步骤s30之后还可以包括:
步骤s40,培养和筛选能正常表达crglut1基因的转基因莱茵衣藻。
以上步骤具体在实施中,可以采用本发明优选方式进行细节操作;其中,
步骤s10的扩增基于后续优选采用的phr13载体质粒具有的酶切位点,扩增过程中优选采用以下引物进行。引物包括具有如下核苷酸序列的上游引物和下游引物:
上游引物:5’-cataatggagccgtgagcaaga-3’;
下游引物:5’-ggatccgggtgctgactctcaagtttgag-3’;
以上下划线部分cata,ggatcc分别为ndei、bamhi酶切位点,用以上引物进行pcr反应扩增目的基因的同时,引入双酶切的酶切位点,从而便于后续与phr13载体质粒的酶切重组。而pcr扩增的操作过程采用常规的pcr步骤进行即可。
同时步骤s20的对步骤s10的扩增产物和phr13载体双酶切之后、再用dna连接酶连接,即能获得重组表达载体phr13-crglut1。
步骤s30的转化过程中,采用工程藻宿主为转基因受体藻株为细胞壁缺陷型莱茵衣藻chlamydomonasreinhardtiijuv进行,使用tap培养基作为莱茵衣藻的培养基,莱茵衣藻的培养条件为22~25℃,光照强度设置为150±50μe/m2/s,转速设置为100rpm/min,待藻细胞生长至对数生长期,细胞密度达到5×106cells/ml时,对藻细胞采用珠磨法进行遗传转化,选择抗生素以及培养基内添加10g/l的葡萄糖作为唯一碳源进行筛选标记。
转化之后,为了验证和筛选能正常表达的转基因莱茵衣藻,步骤s40的培养和验证可以利用pcr检测技术以及荧光定量pcr技术对转基因藻株中crglut1基因鉴定以及表达量的进一步分析。比如,通过筛选培养获得转基因藻株后,利用western-blot技术对crglut1基因所表达的glut1蛋白进行鉴定分析来确定目的基因的表达情况。
还可以采用葡萄糖荧光衍生物2-nbdg(2-(n-7-硝基-2,1,3-苯并恶二唑-4-氨基)-2-脱氧-d-葡萄糖)作为荧光标记物来监测藻细胞对葡萄糖的摄取,检测转基因藻利用葡萄糖能力;具体利用其在485/535nm表现出最大激发和最大发射波长检测藻细胞中2-nbdg水平,从而从功能上印证crglut1基因所表达的蛋白是否能转运外界葡萄糖,来确定表达的蛋白是否为目的蛋白。
进一步还可以对转基因藻进行异养条件下生物量的检测,将其接种于以葡萄糖为单一碳源的tap液体培养基中,调整藻细胞初始浓度至1.0×105cells/ml,分别于黑暗以及光照下培养,培养温度为22℃,120rpm进行培养设置对照组,每组设置3个平行样,每隔24h取一次藻液,在鲁格固定液处理后利用血球计数板对各个浓度下藻液进行计数,绘制细胞密度曲线。结果表明所获得转基因藻能够高效表达葡萄糖转运蛋白,有翻译后的修饰作用和生物活性,其藻细胞能够转运胞外葡萄糖,实现了黑暗条件下藻细胞的异养生长,加速兼养条件下藻细胞生物量的增长。
本发明进一步还提出将以上能成功表达crglut1基因的转基因藻在制备保健食品、异养生物反应器、医药、水产养殖饵料或者动物饲料添加剂中的应用。其表达的具有良好生物活性的目的蛋白glut1,作为医疗、免疫、活性添加成分于饵料/饲料、都具有非常良好的应用前景和价值。
为使本发明上述表达crglut1基因的转基因藻的制备细节更利于本领域技术人员的理解和实施,以及在靶蛋白glut1表达情况的进步性效果,以下通过具体的实施例来对本案的上述内容进行举例说明。
实施例1
s10,合成具有序列表seq.id.no.1的核苷酸序列的crglut1表达基因:由广州艾基生物有限公司进行人工合成,并将合成的基因合成到puc19质粒,构建好的含有目的基因的载体将其名为puc19-crglut1。
s20,重组表达载体phr13-crglut1的制备:
s21,以puc19-crglut1质粒为模板,设计特异性引物并在上下游引物两端分别加上ndei和bamhi酶切位点,对crglut1目的片段进行pcr扩增,扩增出1479bp的crglut1基因片段;其中,引物即为以上所描述的上游引物和下游引物;
上游引物:5’-cataatggagccgtgagcaaga-3’;
下游引物:5’-ggatccgggtgctgactctcaagtttgag-3’;
扩增的过程常用通常的pcr过程进行:94℃预变性5min,94℃变性30s,60℃复性30s,72℃延伸2min,32个循环后,72℃延伸10min。pcr反应程序结束后,分别取5μl反应物进行电泳检测,反应产物-20℃保存;pcr产物用1%的琼脂糖凝胶电泳检测,观察pcr结果,然后将阳性产物置于-20℃保存。其中,该步骤扩增的pcr产物电泳图见图3;图3中条带m为markerdl-5000,条带1为crglut1基因pcr产物。
s22,将步骤s21扩增的阳性产物、以及phr13载体分别用ndei和bamhi核酸内切酶进行双酶切;双酶切反应条件为37℃,20min,80℃热失活5min。使用切胶回收试剂盒分别回收线性大片段phr13和crglut1片段,并用t4连接酶22°将crglut1片段连接到的phr13对应酶切位点上。反应30min后,采用热激法将连接产物转入大肠杆菌中,且将转化后的大肠杆菌涂布于含有相应抗生素的lb平板上。过夜培养后,对所获得的单克隆菌株提取质粒,进行菌落pcr扩增、双酶切验证和测序验证。pcr核酸凝胶电泳结果如图4,双酶切验证的结果如图5、以及测序结果均表明crglut1片段成功连接到了phr13载体上,获得crglut1基因的表达载体phr13-crglut1。其中,图4中电泳结果条带m为markerdl-5000,条带1-4为phr13-crglut1的pcr扩增产物;图5中,条带m为markerdl-5000,条带1为phr13-crglut1双酶切产物。
s30,将步骤s22扩增的表达载体phr13-crglut1与莱茵衣藻受体进行遗传转化:
s31,受体藻珠的选择和培养:选择细胞壁缺陷型莱茵衣藻(chlamydomonasreinhardtii,juv,购自美国杜克大学衣藻遗传中心)。使用tap培养基作为转基因藻株的培养基,其配制方法如下:10mltris-acetate贮存液(242gtris、100ml冰乙酸,溶于水中,定容至1l),10mlphosphatebuffer贮存液(11.92gk2hpo4、6.05gkh2po4,溶于水中定容至1l),10mlbeijerinck’ssolution贮存液(40gnh4cl、10gmgso4·7h20、5gcacl2·2h20、溶于水中定容至1l),1mltrace微量元素混合液(50gna2edta、1.1g(nh4)6mo7o24、1.57gcuso4·5h2o、1.61gcocl2·6h2o、4.99gfeso4·7h2o、22gznso4·7h2o、5.6gmncl2·4h2o、11.4gh3bo3定容至1l去离子水中),将上述溶液混合后溶解于929ml的去离子水中。莱茵衣藻培养条件为22-25℃,光照强度为100μe/m2/s。静置培养至对数中期,当藻细胞的浓度为1-2×106cell/ml时进行以下遗传转化步骤。
s32,在该步骤中采用“珠磨法”进行遗传转化:
1)将细胞壁缺陷性莱茵衣藻juv接种于新鲜tap培养基中进行连续光照培养,待细胞生长至对数期(细胞数为1~2×106cells/ml),取50ml藻液,5000rpm/min常温离心10min,弃去上清液,收集藻细胞;
2)用无菌1mltap培养液重悬藻细胞,调整藻细胞浓度至2×108cells/ml;
3)加300μl藻细胞悬浮液至1.5mlep管里,ep管中有中备好的玻璃珠(0.3mm)分别加入1μg酶切(kpni)成线状并纯化好的phr13-crglut1质粒;
4)把加入了藻细胞/外源dna/玻璃珠混合物的1.5mlep管在振荡器上以最高速充分振荡25s;
5)取10ml新鲜tap培养基到50ml离心管中,把ep管中混合液转移到离心管中轻柔混匀,置于摇床上弱光100rpm,22℃培养过夜,使衣藻细胞恢复;
6)次日,3000rpm/min常温离心5min离心管收集衣藻细胞,弃去上清液;
7)用500μl新鲜tap培养液轻柔重悬细胞,轻柔混匀后平铺在碳源为10g/l的葡萄糖的tap固体平板上(含10μg/ml的潮霉素)静置平板30min后置于22℃光照培养箱中倒置培养约2周长出绿色的单克隆。
s40,转基因藻的筛选:基于phr13-crglut1重组表达载体整合了相应的抗性基因,如hyg基因具有潮霉素抗性,因此用含有10μg/ml潮霉素的抗性平板可以初步筛选出转化成功的单克隆藻株。挑取筛选平板上新生长的单克隆衣藻,于新的筛选tap固体平板上划线扩大培养。待其生长形成规模后,挑取各个克隆藻体入50ml液体tap培养基中扩大培养,在光照条件下培养至对数生长期,收集藻细胞,通过基因组pcr鉴定、qrt-pcr检测、westernblot检测等进行鉴定。具体的鉴定步骤详细包括如下。
s41,转基因藻基因组pcr鉴定:
使用universaldnaextractionkit(takara)提取转基因藻的总dna,提取方法参照说明书。使用crglut1基因引物进行pcr验证,引物及扩增程序同采用与步骤s21中目的基因扩增时相同进行,其基因组pcr鉴定如图6所示。图6中泳道1为转化前重组质粒阳性对照;泳道2为野生型莱茵衣藻阴性对照;泳道3-11为转基因藻基因组pcr产物;从图6所体现的pcr产物的凝胶电泳结果、以及辅助测序结果均表明重组载体phr13-crglut1成功转入到莱茵衣藻基因组中。
s42,qrt-pcr检测crglut1基因表达量:
采用rnafast200-总rna极速抽提试剂盒提取培养至对数后期的野生型莱茵衣藻和转基因藻的总rna,具体操作方法如下:
(1)取10ml衣藻细胞培养液,4℃,10,000rpm离心3min,弃上清收集藻细胞;
(2)加入1ml1%depc水重悬细胞,室温13,000rpm离心1min,弃上清;再加入100μl1%depc水重悬细胞;
(3)加入ra2液500μl,充分颠倒混匀1min;
(4)将样品裂解液吸入内套管中,室温13,000rpm离心1min;
(5)弃去外套管中的液体,内套管中加入500μl洗液,室温13,000rpm离心1min;重复此步骤一次;
(6)弃去外套管中的液体,不加洗液,室温13,000rpm离心1min;
(7)将内套管加在另一新的1.5ml离心管中,在膜的中央加入25μl1%depc水,室温静置1min,13,000rpm离心1min,获得总rna;
(8)取1μl总rna进行1%琼脂糖凝胶电泳分析rna的完整性。
采用rtreagentkitwithgdnaeraser(takara)进行cdna的合成,合成方法参照说明书。采用primer5.0软件设计引物qcrglut1-5f、qcrglut1-5r作为crglut1基因的mrna荧光定量pcr引物。以引物actin-f、actin-r作为检测莱茵衣藻mrna表达的内参基因。使用sybrgreen荧光定量试剂盒进行荧光定量pcr反应,扩增程序如下:95℃预变性30s;95℃变性5s,60℃复性延伸30s,40个循环;测量溶解曲线时,增加步骤:95℃变性15s,60℃复性1min,95℃变性15s。每个样品均设3个重复,在appliedbiosystems7300荧光定量pcr仪上进行定量检测。
所用引物如下:
actin-f:5’-acccgtgctgctgactg-3’;
actin-r:5’-acgttgaaggtctcgaaca-3’;
qcrglut1-5f:5’-cggctccttacaattcggcta-3’;
qcrglut1-5r:5’-ggtgggcagtatggactctcc-3’;
最终qrt-pcr检测结果如图7所示,图7中wt为野生型莱茵衣藻,h5、h9、h11为转crglut1基因衣藻;从图7的结果可以看出,h5、h9、h11转基因藻株crglut1基因表达量相对于野生型藻株分别提高了14.18倍、25.66倍和77.24倍,说明crglut1基因在莱茵衣藻中具有转录活性。
s43,转基因藻的westernblot分析:
使用plantproteinextractionkit(solarbio)提取转基因藻总蛋白,具体操作步骤详见说明书。使用glut1单克隆抗体作为一抗,碱性磷酸酶标记的羊抗兔igg作为二抗,进行免疫印记,检测转基因藻中glut1蛋白的表达。具体方法如下:
(1)取藻细胞的总蛋白以20μg总量18%tris-tricinepage电泳分离,在200v下恒压电泳约2小时,直染料前端抵达凝胶底部;
(2)卸下分离胶,将凝胶浸泡在电转移缓冲液(39mmglycine,48mmtris,0.037%(w/v)sds,10%(v/v)甲醇)中10min,将pvdf浸泡于甲醇溶液中1-2min;
(3)裁剪6张适当大小的滤纸和1张pvdf膜;将滤纸和pvdf膜浸泡在电转移缓冲液中平衡20min;
(4)安装电转移装置,从负极到正极方向依次为:黑色夹板-5层滤纸-凝胶-pvdf膜-5层滤纸-白色夹板;
(5)接通电源,恒流200ma,15min;同时,将电转移仪置于冰浴中;
(6)取出膜,用洗涤缓冲液tbst洗涤3次;
(7)将膜置于20ml3%bsa/tbst封闭液中,4℃,过夜;
(8)按1:500的比例用封闭液稀释glut1单克隆抗体,将膜从封闭液中取出,置于杂交管中,加入稀释的一抗,37℃,杂交1h;
(9)用tbst洗涤杂交膜10min;重复3次;
(10)按1:1000的比例用封闭液稀释碱性磷酸酶标记的羊抗兔igg二抗,将膜置于杂交管中,加入稀释的二抗;37℃,杂交1h;
(11)用tbst洗涤杂交膜10min;重复3次;
(12)将膜置于暗处,加入适量的碱性磷酸酶底物bcip/nbt进行显色;
(13)加入去离子水终止反应。
杂交结果如图8所示,图8中wt为野生型莱茵衣藻总蛋白,h5、h9、h11为转crglut1基因衣藻总蛋白;从图8中可以看出实验组h5、h9、h11出现一条明显的杂交条带,而且空白对照没有出现相应的杂交条带,说明crglut1基因在莱茵衣藻中能够表达出分子量大小约为43kda的目的蛋白。而空白对照则没有表达出相应的石斑鱼抗菌肽piscidin,该结果说明crglut1表达载体在莱茵衣藻细胞内成功表达glut1蛋白,且能够与glut1单克隆一抗进行杂交,实现了首次在莱茵衣藻中表达glut1蛋白。
s44,转基因藻利用葡萄糖能力的检测:
葡萄糖荧光衍生物(2-nbdg)为荧光d-葡萄糖同系物2-(n-7-硝基-2,1,3-苯并恶二唑-4-氨基)-2-脱氧-d-葡萄糖,一种脱氧葡萄糖类似物,常作为荧光标记物来监测活细胞对葡萄糖的摄取,能够利用其在485/535nm表现出最大激发和最大发射波长从而检测藻细胞中2-nbdg水平。
使用caymanglucoseuptakecell-basedassaykit对转化株进行葡萄糖荧光衍生物(2-nbdg)转运检测。
1)取1ml对数生长前期的转化株,5000rpm/min,5min室温离心培养液,去上清,加入1ml无碳源tap液体培养基,并加入15μl浓度为10mg/ml的2-nbdg,黑暗过夜培养;
2)5000rpm/min,5min室温离心培养液,去除上清液;
3)每个样品加200μlcell-basedassaybuffer,轻柔混匀再次离心5min,5000rpm/min,5min室温离心培养液,去除上清液;
4)每个样品加入100μlcell-basedassaybuffer后,于台式钙流工作站检测荧光强度(激发/发射=485/535nm)。
检测结果如图9所示,图9中wt为野生型莱茵衣藻,z1、z2、h9、h11为转crglut1基因衣藻,**表示与野生型莱茵衣藻藻细胞内2-nbdg含量相比产生显著性差异(0.01<p<0.05),***表示与与野生型莱茵衣藻藻细胞内2-nbdg含量相比产生极显著性差异(p<0.01)。
根据图9的结果可以看出,转化株均有对应荧光发出,对照组荧光强度最低。与对照组相比,h9转化株荧光强度最高,为对照组的10.94倍,其余转化株与对照组相比分别是z1转化株为7.38倍,z2转化株为5.08倍,h11转化株为10.01倍,这表明藻细胞中含有葡萄糖荧光衍生物2-nbdg,说明葡萄糖荧光衍生物2-nbdg在黑暗培养过程中均被转化株藻细胞吸收,葡萄糖转运glut1蛋白具有活性且能够将外界葡萄糖转运至藻细胞中。
s45,转基因藻异养与兼养条件下生物量的检测:
将转基因藻接种于以葡萄糖为单一碳源的tap液体培养基中,调整藻细胞初始浓度至1.0×105cells/ml,分别于黑暗以及光照下培养,培养温度为25℃,120rpm进行培养设置对照组,每组设置3个平行样,每隔24h取一次藻液,在鲁格固定液处理后利用血球计数板对各个浓度下藻液进行计数,绘制细胞密度曲线。
黑暗异养条件下结果如图10所示,图10中中wt为野生型莱茵衣藻,z1、z2、h9、h11为转crglut1基因衣藻;根据图10的结果可以看出,在黑暗条件以及葡萄糖作为唯一碳源的情况下,转化株生物量明显高对照组,对照组的生长几乎停滞,其中转化株h9,h11生物量比z1,z2生物量高出约30%,达到5×105cells/ml。此结果说明,转基因藻在黑暗条件下能利用葡萄糖进行生长。
兼养条件下的结果如图11所示,图11中中wt为野生型莱茵衣藻,z1、z2、h9、h11为转crglut1基因衣藻;根据图11的结果可以看出,在光照条件以及葡萄糖作为唯一碳源的情况下,转化株生物量与对照组有明显增加,转化株z1,z2,h9,h11分别增加了45%,55%,67%以及73%,且h9转化株比其他转化株更早到达平台期,生长速度更快。
结果表明所获得转基因藻能够高效表达葡萄糖转运蛋白,有翻译后的修饰作用和生物活性,其藻细胞能够转运胞外葡萄糖,实现了黑暗条件下藻细胞的异养生长,加速兼养条件下藻细胞生物量的增长。
以上仅为本发明的优选实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
序列表
<110>深圳大学
<120>crglut1表达基因、表达载体、莱茵衣藻工程藻及制备方法和应用
<160>3
<170>patentinversion3.3
<210>1
<211>1479
<212>dna
<213>人工序列
<220>
<223>crglut1表达基因
<400>1
atggagccgtcgagcaagaagctgacgggacgtctgatgctcgccgttggcggcgcggtc60
ctcggctccttacaattcggctacaacaccggcgtgattaacgccccgcagaaggtgatc120
gaggagttctacaatcagacgtgggtgcaccgctacggagagtccatactgcccaccacc180
ctgaccaccctgtggtccctgtccgtggccattttctccgtgggcggcatgatcggcagt240
ttctcggtgggcctattcgtgaaccgcttcggccgtcgcaacagcatgctgatgatgaac300
ctgctggcctttgtcagtgccgtgctgatgggcttctcgaagctgggaaaatcattcgag360
atgctgatcctcggccgcttcatcatcggtgtgtactgcggtctgacaaccggcttcgtg420
ccgatgtacgtgggcgaggtctcgcccacggcgctgcgcggcgcgctgggcacgctgcac480
caactcggcatcgtcgtgggcatcctgatcgcccaggttttcggactggactccatcatg540
gggaacaaggacttgtggcccctgctgctgagcatcattttcattcccgccctccttcag600
tgcatcgtgctgccgttctgccccgagtcccctcgcttcctgctgataaaccgcaacgag660
gagaaccgcgccaagtccgtgctgaagaagctgcgcgggactgctgacgtcacccacgac720
ttacaagagatgaaggaggagtcaaggcagatgatgcgcgagaagaaggtgacgatcctg780
gagctgtttcgctcccccgcctaccgccagcctatcctgatcgcggtcgtgttacaactg840
agccagcagctcagcggtataaacgcggtgttctactacagcacgtctatcttcgagaag900
gcaggcgtgcagcagccggtgtacgccaccatcggcagtggtatcgtcaataccgccttt960
acggtagtgagcctcttcgtggtcgagcgcgcgggtcgccggacattgcacctgatcggc1020
ctggccggcatggccggctgcgccattctgatgactattgcgctggcccttctggagcag1080
ctgccatggatgtcgtacctgtccatcgtcgccattttcggcttcgtggcattctttgag1140
gtgggcccgggacccatcccctggttcatcgtggccgagctgttcagccagggtccccgg1200
cccgccgcgatcgccgtcgccgggttctcaaactggacgtcaaacttcatcgtgggcatg1260
tgctttcaatacgtcgagcagctgtgcggcccctacgtgttcatcattttcacggtcctg1320
ctcgtgttgttctttatcttcacgtattttaaggtgcccgagacgaagggcaggacgttc1380
gacgagatcgctagcggcttccgccagggcggcgcctcccagtcggacaagacccctgag1440
gagctgttccacccgctgggtgctgactctcaagtttga1479
<210>2
<211>22
<212>dna
<213>人工序列
<220>
<223>上游引物
<400>2
cataatggagccgtgagcaaga22
<210>3
<211>20
<212>dna
<213>人工序列
<220>
<223>下游引物
<400>3
ggatccgggtgctgactctcaagtttgag29
- 还没有人留言评论。精彩留言会获得点赞!