一种治疗卵巢癌药物Rucaparib中间体的制备方法与流程
本发明属于医药中间体的制备领域,具体涉及一种治疗卵巢癌药物rucaparib中间体的制备方法。
背景技术:
rucaparib(瑞卡帕布)是一种聚腺苷二磷酸核糖聚合酶(parp)抑制剂,作为单药治疗brca(breastcancersusceptibilitygene乳腺癌易感基因突变)的晚期卵巢癌。
rucaparib化学名为:8-氟-2-{4-[(甲氨基)甲基)]苯基}-1,3,4,5-四氢-6h-氮杂卓并[5,4,3-cd]吲哚-6-酮磷酸盐,分子式为:c19h18fn3o·h3po4,cas:459868-92-9,其结构式如下:
文献(oprd,16,12,(2012),p.1897–1904)报道了rucaparib的合成方法,合成路线如下:
其中,中间体v(化学名:8-氟-1,3,4,5-四氢-2-[4-[(甲基氨基)甲基]苯基]-6h-吡咯并[4,3,2-ef][2]苯并氮杂-6-酮),是一种制备rucaparib的重要中间体。其结构式如下:
目前,中间体v的合成路线主要有:
1、文献(oprd,16,12,(2012),p.1897–1904),合成路线如下所示:
该制备方法较为繁琐,需要用到易爆危险的硝化反应,大量的废酸需要处理,需要多次氢气还原,需过柱纯化,总收率较低。
2、文献(oprd,16,12,(2012),p.1897–1904),合成路线如下所示:
该制备方法所用原料价格高,难以获得,不适用于工业化生产。
3、文献(耿元硕等,精细化工中间体,2012,42(5)),合成路线如下所示:
该制备方法反应步骤多,使用大量硝酸、硫酸、三氯氧磷,收率低,成本高,不适合于工业化生产。
因此,研究中间体v的新合成路线及工艺,对rucaparib的合成具有重要意义。
技术实现要素:
发明目的:本发明目的在于针对现有技术中rucaparib中间体(中间体v)的制备方法还存在条件苛刻、工艺复杂、反应时间过长以及收率偏低的缺陷,提供一种治疗卵巢癌药物rucaparib中间体的制备方法。本发明的制备方法安全环保、原料易得、成本低、反应收率高,适合于工业化生产。
技术方案:本发明的目的通过下述技术方案实现:
一种治疗卵巢癌药物rucaparib中间体的制备方法,包括以下步骤:
(1)式iii化合物与氨基乙醛缩醇化合物在缩合剂或羧酸活化剂作用下发生缩合反应,得到式iv化合物:
(2)式iv化合物在酸和还原剂的作用下,得到式v化合物,即所述药物rucaparib的中间体:
优选地,所述的治疗卵巢癌药物rucaparib中间体的制备方法,包括以下步骤:
(1)将式iii化合物和氨基乙醛缩醇化合物与有机溶剂a混合,加入缩合剂或羧酸活化剂,0℃至溶剂回流温度下反应1-20h;反应完毕后,往反应液中加入水,所得产物可从反应液中析出,过滤析出的固体,重结晶得式iv化合物;所述式iii化合物、缩合剂或羧酸活化剂、氨基乙醛缩醇化合物的摩尔比为1:1-5:1-2。
所述有机溶剂a优选乙腈、甲苯、二氯甲烷、乙酸乙酯、dmf等,这取决于缩合反应选用的缩合剂或羧酸活化剂;反应温度取决于所选用的缩合剂或羧酸活化剂,一般为0℃至溶剂回流温度;反应时间一般为1-20h,这取决于反应体系中混合物的组成。反应完毕后,往反应液中加入水,过滤析出的固体。如析出固体较少,也可用乙酸乙酯、二氯甲烷等溶剂萃取后浓缩得到固体。重结晶溶剂优选:二氯己烷/正己烷混合液。
(2)将式iv化合物和还原剂与有机溶剂b混合,降温至0-10℃,加入酸,0℃至溶剂回流温度下反应8-12h,过滤析出的固体,洗涤,干燥得式v化合物;所述式iv化合物、还原剂、酸的摩尔比为1:1-10:1-5。
所述有机溶剂b优选二氯甲烷、氯仿、甲苯等溶剂;反应温度一般为0℃至溶剂回流温度,优选0-30℃。反应完毕后,所得产物可从反应液中析出,过滤,真空干燥即得。
优选地,所述步骤(1)中,缩合剂选自羰基二咪唑cdi、碳二亚胺类缩合剂或鎓盐类缩合剂;羧酸活化剂选自氯甲酸甲酯、二氯亚砜、磺酰氯、草酰氯或混合酸酐中的一种或几种;氨基乙醛缩醇化合物选自氨基乙醛缩二甲醇或氨基乙醛缩二乙醇。
进一步优选地,所述碳二亚胺类缩合剂选自1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐edci或二环己基碳二亚胺dcc。
进一步优选地,所述鎓盐类缩合剂选自2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯hatu、苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐hbtu或苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐bop中任一种。
优选地,所述步骤(2)中,酸选自三氟乙酸、三氟化硼乙醚或三硝基苯磺酸中任一种;还原剂选自三乙基硅烷、三正丁基硅烷、三苯基硅烷或三异丙基硅烷中任一种。
优选地,所述式iii化合物的制备包括以下步骤:
(1)式i化合物与氰化试剂反应,得到式ii化合物:
(2)式ii化合物在碱性条件或酸性条件下水解,得到式iii化合物:
进一步优选地,所述步骤(1)中,将式i化合物在有机溶剂c中,与氰化试剂在催化剂作用下反应,反应温度20-200℃,反应时间1-20h;所述氰化试剂选自氰化亚铜或氰化锌;所述有机溶剂c选自dmf,dma,dmso或nmp中任一种;所述催化剂选自pd或ni催化剂;所述式i化合物与氰化试剂的摩尔比为1:0.5~10。
步骤(1)将式i化合物在有机溶剂中与氰化试剂反应,将芳基溴变为氰基。所述催化剂优选pd和ni催化剂,例如:pd(dppf)cl2,pd(oac)2或pd2(dba)3;反应温度一般为20-200℃,优选80-150℃;反应时间一般为1-20h,这取决于反应体系中混合物的组成和所选择的温度范围。反应完毕后,所得混合物可通过一层硅藻土过滤,滤液进行萃取处理,在真空中浓缩溶剂后,即得式ii化合物。
进一步优选地,所述步骤(2)中,碱性条件或酸性条件下水解反应温度为80-150℃,反应时间为1-20h;对于1mol的式ii化合物,使用1-10mol的无机碱或无机酸。
式ii化合物水解为式iii化合物,这可通过技术人员已知的用于水解芳香氰基的方法来进行,例如:michaelb.smith&jerrymarch,"march'sadvancedorganicchemistry:reactions,mechanisms,andstructure"(6thedition),2007,wileyisbn:0471720917,或richardc.larock,"comprehensiveorganictransformations:aguidetofunctionalgrouppreparations"(2ndedition),1999,wiley,isbn:0471190314和其中所引用的文献中所示的那样。例如:在碱存在下水解,对于1mol的待反应的式ii化合物,使用1-10当量的碱;碱优选氢氧化钠或氢氧化钾;溶剂优选水或与水互溶的有机溶剂,例如:乙二醇、甲醇、乙醇、dmf、dmso或nmp;反应温度优选80-150℃;反应时间一般为1-20h,这取决于反应体系中混合物的组成和所选择的温度范围。反应完毕所得混合物可通过调节ph至1-4,过滤析出的固体,在真空中干燥后得到。例如:在酸存在下水解,可选的酸优选盐酸、磷酸和硫酸;对于1mol的待反应的式ii化合物,使用1-10当量的酸,优选用酸做溶剂;反应温度优选80-150℃;反应时间一般为1-20h,这取决于反应体系中混合物的组成和所选择的温度范围。反应完毕所得混合物可通过加入水,过滤析出的固体,在真空中干燥得到。
优选地,所述的治疗卵巢癌药物rucaparib中间体的制备方法,包括以下步骤:
以式i化合物为起始原料,与氰化试剂反应,得到式ii化合物;式ii化合物在碱性条件或酸性条件下水解,得到式iii化合物;式iii化合物与氨基乙醛缩醇化合物在缩合剂或羧酸活化剂作用下发生缩合反应,得到式iv化合物;式iv化合物在酸和还原剂的作用下,得到式v化合物,即所述药物rucaparib的中间体。
其反应式为:
本发明的有益效果在于:
(1)本发明的制备方法,路线设计合理,避免了硝化反应,避免使用大量强酸,保证安全生产,安全环保;反应条件温和易控,有效缩短了反应路线,缩短了反应周期,降低了生产成本,适合于工业化生产。
(2)本发明的制备方法,采用的原料易得,成本低,收率高,便于工业化生产。
附图说明
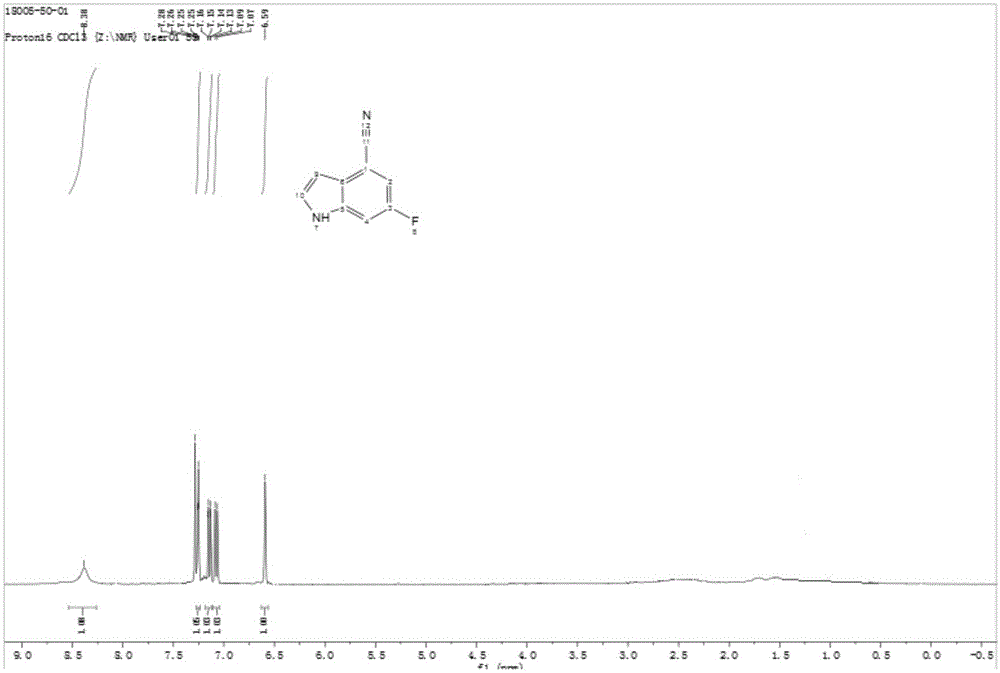
图1为实施例1式ii化合物的hnmr谱图;
图2为实施例1式iii化合物的hnmr谱图;
图3为实施例1式iv化合物的hnmr谱图;
图4为实施例1式v化合物的hnmr谱图。
具体实施方式
下面通过具体实施例对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
实施例1
a)6-氟-4-氰基-1h-吲哚
将式i化合物(460g),氰化亚铜(290g)和n,n-二甲基甲酰胺(2.3l)混合,氮气置换3次,加热至150℃搅拌6小时。反应完毕后,冷却至室温。
往反应液中加入水(7l)和乙酸乙酯(2l),通过硅藻土过滤,滤液用乙酸乙酯(2l*2)萃取两次,有机层用无水硫酸钠干燥,用活性炭脱色,浓缩至干得灰色固体(式ii化合物)310g,纯度:97.51%,收率:90%。
式ii化合物的hnmr谱图见附图1。
hnmr(400mhz,cdcl3):δ9.38(brs,1h),7.25(t,j=4.0hz,1h),7.15(dd,j=8.0,4.0hz,1h),7.08(d,j=8.0hz,1h),6.59(t,j=4.0hz,1h)。
b)6-氟-4-羧基-1h-吲哚
将式ii化合物(290g)、氢氧化钠(580g)和水(1.2l)混合,加热至120℃搅拌8小时。反应完毕后,冷却至室温。
往反应液中加入水(3l),用乙酸乙酯(500ml)萃取。取水层,水层用浓盐酸调节ph至2-3,固体析出,过滤析出的固体。加入异丙醇打浆,过滤,干燥得灰色固体(式iii化合物)240g,纯度:97.58%,收率:74%。
式iii化合物的hnmr谱图见附图2。
hnmr(400mhz,dmso-d6):δ12.94(brs,1h),11.46(brs,1h),7.50(s,1h),7.46(d,j=8.0hz,2h),7.01(s,1h)。
c)n-(2,2-二甲氧基乙基)-6-氟-1h-吲哚-4-酰胺
将式iii化合物(220g)与乙腈(2.2l)混合,加入n,n’-羰基二咪唑(210g),加热回流1小时,加入氨基乙醛缩二甲醇(140g),加热回流3小时。反应完毕后,往反应液中加入水,过滤析出的固体,50℃真空干燥。所得固体用二氯甲烷/正己烷重结晶得淡黄色固体(式iv化合物)280g,纯度:98.64%,收率:88%。
式iv化合物的hnmr谱图见附图3。
hnmr(400mhz,cdcl3):δ9.17(brs,1h),8.10(brs,1h),7.40(t,j=4.0hz,1h),7.13(dd,j=8.0,4.0hz,1h),6.84(d,j=8.0hz,1h),6.54(t,j=4.0hz,1h),4.88(t,j=4.0hz,1h),3.51(s,6h),3.42(m,2h)。
d)8-氟-1,3,4,5-四氢-2-[4-[(甲基氨基)甲基]苯基]-6h-吡咯并[4,3,2-ef][2]苯并氮杂-6-酮
将式iv化合物(250g)和二氯甲烷(2l)混合,加入三乙基硅烷(220g),降温至0℃,滴加三氟乙酸(210g)的二氯甲烷溶液(1l)。滴加完毕后,在20℃搅拌8小时。
过滤析出的固体,用二氯甲烷洗涤,于40℃真空干燥得淡黄色固体(式v化合物)140g,纯度:98.62%,收率:75%。
式v化合物的hnmr谱图见附图4。
hnmr(400mhz,dmso-d6):δ11.13(brs,1h),8.11(t,j=5.5hz,1h),7.37(dd,j=9.3,2.4hz,1h),7.30(dd,j=9.3,2.4hz,1h),7.22(brs,1h),3.37(q,j=5.5hz,2h),2.86-2.88(m,2h)。
实施例2
a)6-氟-4-氰基-1h-吲哚
将式i化合物(360g),氰化锌(400g)和二甲亚砜(1.8l)混合,加入pd(dppf)cl2(18g),氮气置换3次。加热至120℃搅拌8小时。反应完毕后,冷却至室温。
往反应液中加入水(5.4l)和乙酸乙酯(2l),通过硅藻土过滤,滤液用乙酸乙酯(2l*2)萃取两次,有机层用无水硫酸钠干燥,活性炭脱色,浓缩至干得灰色固体(式ii化合物)250g,纯度:95.87%,收率:93%。
b)6-氟-4-羧基-1h-吲哚
将式ii化合物(240g)和浓盐酸(1.2l)加入压力反应釜,加热至120℃搅拌10小时。反应完毕后,冷却至室温。
往反应液中加入水(3.6l),过滤析出的固体,用水洗涤,鼓风干燥得灰色固体(式iii化合物)191g,纯度:97.96%,收率:71%。
c)n-(2,2-二甲氧基乙基)-6-氟-1h-吲哚-4-酰胺
将式iii化合物(180g)溶于二氯甲烷(1.5l),降温至0℃,滴加草酰氯(140g),滴加完毕,保温搅拌2小时。将反应液滴加到氨基乙醛缩二甲醇(7.5g)和三乙胺(142g)的二氯甲烷溶液(1.5l)中,滴加完毕后,保温搅拌2小时。反应完毕后,往反应液中加入水(3l),分层,用二氯甲烷(1l*2)萃取两次,浓缩至少量,用乙酸乙酯/正己烷重结晶得(式iv化合物)160g,纯度:98.41%,收率:89%。
d)8-氟-1,3,4,5-四氢-2-[4-[(甲基氨基)甲基]苯基]-6h-吡咯并[4,3,2-ef][2]苯并氮杂-6-酮
将式iv化合物(150g)溶于二氯甲烷(1.5l),加入三乙基硅烷(131g),降温至5℃,滴加三氟化硼乙醚(159g)。滴加完毕后,在30℃搅拌12小时。
过滤析出的固体,用二氯甲烷洗涤,于40℃真空干燥得淡黄色固体(式v化合物)82g,纯度:98.85%,收率:72%。
实施例3
a)6-氟-4-氰基-1h-吲哚
将式i化合物(750g),氰化亚铜(377g),碘化钾(872g)和n,n’-二甲基甲酰胺(3.7l)混合,氮气置换3次。加热至回流搅拌16小时。冷却至室温。
往反应釜中加入水(11l)和乙酸乙酯(3l),通过硅藻土过滤,滤液用乙酸乙酯(3l*2)萃取两次,有机层用无水硫酸钠干燥,用活性炭脱色,浓缩至干得灰色固体(式ii化合物)455g,纯度:98.01%,收率:81%。
b)6-氟-4-羧基-1h-吲哚
将式ii化合物(510g)、氢氧化钾(893g)、乙二醇(1.2l)和水2.4l混合,加热至120℃搅拌20小时。反应完毕后,冷却至室温。
往反应釜中加入水(7l),用甲基叔丁基醚(3l)萃取。取水层,水层用浓盐酸调节ph至2-3,固体析出,过滤析出的固体。干燥得灰色固体(式iii化合物)382g,纯度:97.95%,收率:67%。
c)n-(2,2-二甲氧基乙基)-6-氟-1h-吲哚-4-酰胺
将式iii化合物(200g)和氨基乙醛缩二甲醇(130g)溶于n,n’-二甲基甲酰胺(1.5l)中,加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐edci(322g),室温搅拌5小时。反应完毕后,往反应液中加入水(4.5l),过滤析出的固体,减压干燥得淡黄色固体(式iv化合物)250g,纯度:97.94%,收率:84%。
d)8-氟-1,3,4,5-四氢-2-[4-[(甲基氨基)甲基]苯基]-6h-吡咯并[4,3,2-ef][2]苯并氮杂-6-酮
将式iv化合物(110g)溶于二氯甲烷(1l)中,加入三正丁基硅烷(248g),降温至0℃,滴加三氟乙酸(94g)的二氯甲烷溶液(1l)。滴加完毕后,在20℃搅拌10小时。
过滤析出的固体,用二氯甲烷洗涤,于40℃真空干燥得淡黄色固体(式v化合物)84g,纯度:99.11%,收率:78%。
实施例4
a)6-氟-4-氰基-1h-吲哚
将式i化合物(198g),氰化锌(64g)和n-甲基吡咯烷酮nmp(0.8l)混合,氮气置换2次,加入三(二亚苄基丙酮)二钯pd2(dba)3(10g)和三苯基膦pph3(6g),氮气再置换一次。加热至120℃搅拌12小时。反应完毕后,冷却至室温。
往反应液中加入水(2l)和乙酸乙酯(1l),通过硅藻土过滤,滤液用乙酸乙酯(2l*2)萃取两次,有机层用无水硫酸钠干燥,通过硅胶短柱过滤,滤液浓缩至干得灰色固体(式ii化合物)130g,纯度:98.17%,收率:88%。
b)6-氟-4-羧基-1h-吲哚
将式ii化合物(520g)和浓磷酸(85wt.%inh2o)(1.5l)混合,加热至150℃搅拌2小时。反应完毕后,冷却至室温。
往反应液中加入水(6l),固体析出,过滤析出的固体。加入异丙醇(1l)打浆,过滤,干燥得灰色固体(式iii化合物)390g,纯度:97.78%,收率:67%。
c)n-(2,2-二乙氧基乙基)-6-氟-1h-吲哚-4-酰胺
将式iii化合物(263g)、氨基乙醛缩二甲醇(234g)和n,n’-二甲基甲酰胺(790ml)混合,加入2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯hatu(837g),室温搅拌8小时。反应完毕后,往反应液中加入水(4l),过滤析出的固体,用水洗涤,固体于50℃真空干燥5小时,用二氯甲烷/正己烷重结晶得淡黄色固体(式iv化合物)311g,纯度:98.37%,收率:72%。
d)8-氟-1,3,4,5-四氢-2-[4-[(甲基氨基)甲基]苯基]-6h-吡咯并[4,3,2-ef][2]苯并氮杂-6-酮
将式iv化合物(318g)溶于二氯甲烷(0.8l),加入三正丁基硅烷(650g),降温至0℃,滴加三氟化硼乙醚(460g)的二氯甲烷溶液(1l)。滴加完毕后,于20℃搅拌12小时。
过滤析出的固体,用二氯甲烷洗涤,于50℃真空干燥得淡黄色固体(式v化合物)152g,纯度:98.73%,收率:69%。
如上所述,尽管参照特定的优选实施例已经表示和表述了本发明,但其不得解释为对本发明自身的限制。在不脱离所附权利要求定义的本发明的精神和范围前提下,可对其在形式上和细节上作出各种变化。
- 还没有人留言评论。精彩留言会获得点赞!