一种多齿配体三联吡啶双乙酸及其制备方法与流程
本发明涉及一种发光材料领域,特别是一种多齿配体三联吡啶双乙酸及其制备方法。
背景技术:
将含有3d、4d电子的金属离子以及4f电子的稀土离子通过有机共轭配体桥联形成的配位聚合物,不仅结构新颖奇特,往往还呈现出特殊的光、电、磁以及催化性能。d-f型的异核配合物更是能实现这些性能的复合优化,在新型发光材料领域有着潜在应用前景。在构筑这些配位聚合物的独特结构时,桥联配体的选择至关重要。2,2’:6’,2”-三联吡啶基团依靠{n,n,n}配位,具有良好的配位能力,在光电子领域有着很好的应用[u.s.schubertet.al.,chem.soc.rev.,2011,40,1459-1511]。三联吡啶分子结构中的芳香环便于结构修饰,可在多个位置连接给电子/吸电子基团,或者生物相容性的大分子基团,从而可进一步优化三联吡啶配合物的性能[g.r.newkomeet.al.,chem.soc.rev.,2018,47,3991-4016]。羧酸基团中的两个o原子可以通过单配、双配甚至桥联的方式与金属离子结合,得到多种配位结构共存的配合物[a.a.sidorovet.al.,russ.j.coord.chem.,2016,42,621-634],这些结构的配合物在催化、磁性材料等领域应用广泛。如何将三联吡啶配合物与羧酸配合物的优势结合起来是一个巨大的挑战,目前常见的做法是将两种配体同时与金属离子反应,或者分步反应以实现混配,这些方法的副反应较多,提纯步骤复杂,产率不高。
技术实现要素:
本发明要解决的技术问题是针对现有技术的不足,提出了一种配位能力强,配位方式多样的多齿配体三联吡啶双乙酸。本发明另一要解决的技术问题是提出了一种该多齿配体三联吡啶双乙酸的制备方法。
本发明要解决的技术问题是通过以下技术方案米实现的,一种多齿配体三联吡啶双乙酸,其特点是具有如下结构:
分子式c25h20n4o4,分子量440。
化合物名称:4’-p-n,n-二(羧乙基)胺基苯基-2,2’:6,2”-三联吡啶。
缩写词:tpypdca。
上述多齿配体三联吡啶双乙酸的一种制备方法,其特点是包括如下步骤:
(1)、将苯胺、碳酸钾、碘化钾按照1∶1~3∶1~3的摩尔比配成混合物,在氮气保护下搅拌10~30分钟;
缓慢的将溴乙酸乙酯的乙腈溶液滴加到上述混合物中,其中溴乙酸乙酯与乙腈的体积比为1∶3~5,滴加的量按苯胺与溴乙酸乙酯的摩尔比为1∶2~3.5计量;
在90~120℃下搅拌6~12小时,反应结束后蒸除溶剂,将余渣溶解在二氯甲烷中,并用水洗涤,取下层有机层用无水硫酸钠干燥;过滤后浓缩,然后用中性氧化铝色谱柱提纯,收集淡黄色产物得到p-n,n-二酯基苯胺;
(2)、将p-n,n-二酯基苯胺、三氯氧磷、n,n-二甲基甲酰胺按质量比为1∶1.0~1.2∶1.0~1.5的比例混合后,在-10~10℃条件下搅拌0.5~1小时后升温至80~100℃,反应3~8小时;
冷却至室温,调节ph=7~9;
然后用二氯甲烷萃取,有机层水洗后用无水硫酸钠干燥;过滤后浓缩,然后用中性氧化铝色谱柱提纯,收集淡黄色产物得到p-n,n-二酯基苯甲醛;
(3)、将p-n,n-二酯基苯甲醛、2-乙酰基吡啶溶解于乙醇中,再加入氢氧化钠水溶液进行混和;
其中p-n,n-二酯基苯甲醛、2-乙酰基吡啶、naoh水溶液、乙醇的质量比为1∶1.0~1.5∶1.0~1.3∶4.0~7.0,其中氢氧化钠水溶液的浓度为10mol/l;
室温搅拌2~3小时后,-4℃冷冻0.5~2小时,除去上层液体;室温下,对下层有机物再加入乙醇并搅拌至黑红色油状物全部溶解;
然后再加入naoh水溶液和氨水,其中加入naoh水溶液和氨水的量与上述2-乙酰基吡啶用量三者的质量比为1.0~1.5∶10~15∶1,其中氢氧化钠的水溶液的浓度为10mol/l,氨水的质量百分比浓度为25%;
在80~120℃条件下反应3~8小时,反应完成后,将混合物倒入冷水中冷却,调节ph至3.5~6.5,得到黄色沉淀,离心分离后的固体用乙醇和无水乙醚的混合溶剂洗涤,其中乙醇和无水乙醚的体积比为1∶10~20;过滤后干燥,得到三联吡啶双乙酸。
本发明要解决的技术问题还可以通过以下技术方案来进一步实现,用中性氧化铝色谱柱提纯时使用的洗脱液为石油醚与乙酸乙酯或乙醇的混合液,其中石油醚∶乙酸乙酯或乙醇的体积比为1∶1~1.5。
本发明要解决的技术问题还可以通过以下技术方案来进一步实现,步骤(1)中蒸除溶剂的方法采用减压蒸馏法,其压力为-0.09~-0.1mpa。
本发明通过在苯环的对位上分别连接一个三联吡啶以及一个胺基二乙酸基团,得到多齿配体三联吡啶双乙酸。三联吡啶对大部分d过渡金属,特别是zn(ii)、ru(ii)、ir(iii)、re(i)等离子具有良好亲和性,羧基能和部分d过渡金属离子离子,特别是zn(ii)、sn(iv),以及稀土离子,特别是eu(iii)、tb(iii)配位。通过控制三联吡啶双乙酸与金属离子的反应配比,调节合适的反应条件,如温度、溶剂、反应时间、ph值等,可以便捷的得到多种结构的同核和异核的d-d型或d-f型配位聚合物。
与现有技术相比,本发明的三联吡啶双乙酸的配位能力强,配位方式多样。本发明的三联吡啶双乙酸的制备方法操作简单,易于纯化,产率高。以三联吡啶或羧酸为配体的金属配合物种类繁多,应用广泛。依托本发明的三联吡啶双乙酸的合成方法,并以之为多齿配体与合适的金属离子在特定条件下反应,可获得一系列适宜于发光材料、磁性材料以及催化剂研究的配位聚合物。
附图说明
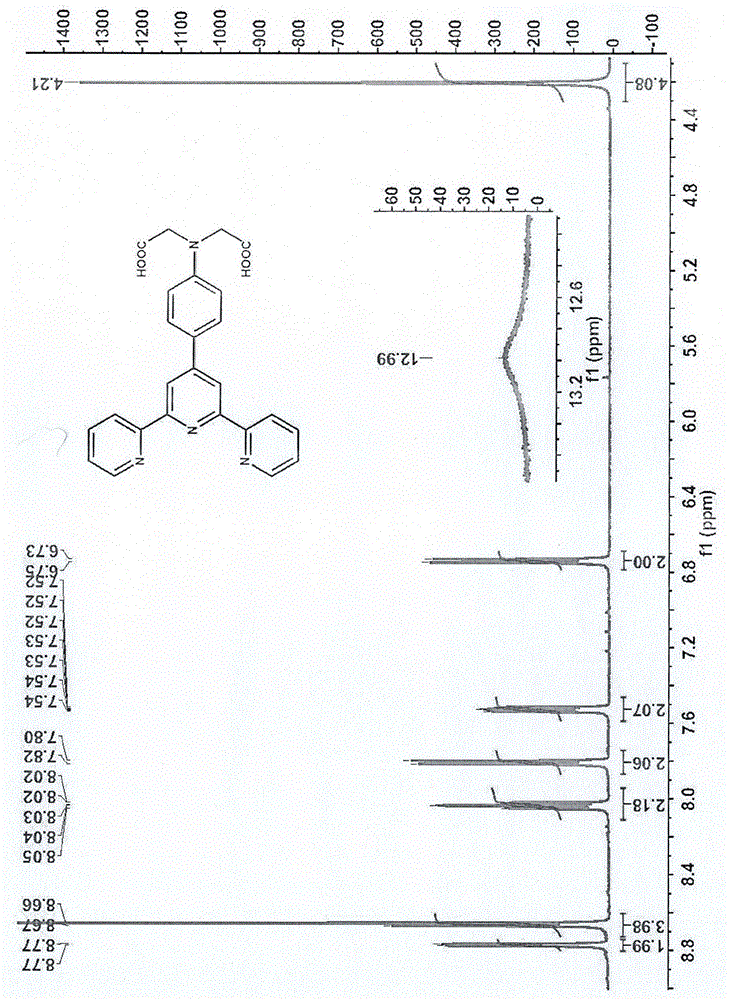
图1为本发明的核磁氢谱;
图2为本发明的电喷雾质谱;
图3为本发明的红外光谱;
图4为本发明的荧光光谱图;
图5为本发明的锌配合物的荧光光谱图;
图6为本发明制备步骤中的合成路线图;
图7为本发明的锌配合物的单晶x-射线衍射图。
具体实施方式
以下进一步描述本发明的具体技术方案,以便于本领域的技术人员进一步地理解本发明,而不构成对其权利的限制。
一种多齿配体三联吡啶双乙酸,其特点是具有如下结构:
分子式c25h20n4o4,分子量440。
化合物名称:4’-p-n,n-二(羧乙基)胺基苯基-2,2’:6,2”-三联吡啶,缩与词:tpypdca。
上述多齿配体三联吡啶双乙酸的一种制备方法,包括如下步骤:
(1)、将苯胺、碳酸钾、碘化钾按照1∶1~3∶1~3的摩尔比配成混合物,在氮气保护下搅拌10~30分钟;
缓慢的将溴乙酸乙酯的乙腈溶液滴加到上述混合物中,其中溴乙酸乙酯与乙腈的体积比为1∶3~5,滴加的量按苯胺与溴乙酸乙酯的摩尔比为1∶2~3.5计量;
在90~120℃下搅拌6~12小时,反应结束后蒸除溶剂,将余渣溶解在二氯甲烷中,并用水洗涤两次,除去上层液,包括反应不完全的无机盐类;
取下层有机层用无水硫酸钠干燥,看不到水层为止,干燥时间为4~6小时;过滤后浓缩,然后用中性氧化铝色谱柱提纯,洗脱液为石油醚∶乙酸乙酯或乙醇=1∶1~1.5,收集淡黄色产物得到p-n,n-二酯基苯胺;
(2)、将p-n,n-二酯基苯胺、三氯氧磷、n,n-二甲基甲酰胺按质量比为1∶1.0~1.2∶1.0~1.5的比例混合后,在-10~10℃条件下搅拌0.5~1小时后升温至80~100℃,反应3~8小时;
冷却至室温,用醋酸钠或碳酸钠或氢氧化钠中的一种或几种调节ph=7~9;
然后用二氯甲烷萃取,有机层水洗后用无水硫酸钠干燥;过滤后浓缩,然后用中性氧化铝色谱柱提纯,洗脱液为石油醚∶乙酸乙酯或乙醇=1∶1~2.5,收集淡黄色产物得到p-n,n-二酯基苯甲醛;
(3)、将p-n,n-二酯基苯甲醛、2-乙酰基吡啶溶解于乙醇中,再加入氢氧化钠水溶液进行混和;
其中p-n,n-二酯基苯甲醛、2-乙酰基吡啶、naoh水溶液、乙醇的质量比为1∶1.0~1.5∶1.0~1.3∶4.0~7.0,其中氢氧化钠水溶液的浓度为10mol/l;
室温搅拌2~3小时后,-4℃冷冻0.5~2小时,除去上层液体;室温下,对下层有机物再加入乙醇并搅拌至黑红色油状物全部溶解;
然后再加入naoh水溶液和氨水,其中加入naoh水溶液和氨水的量与上述2-乙酰基吡啶用量三者的质量比为1.0~1.5∶10~15∶1,其中氢氧化钠的水溶液的浓度为10mol/l,氨水的质量百分比浓度为25%;
在80~120℃条件下反应3~8小时,反应完成后,将混合物倒入冷水中冷却,用稀hcl、稀硫酸或者醋酸中一种或几种调节ph至3.5~6.5,得到黄色沉淀,离心分离后的固体用乙醇和无水乙醚的混合溶剂洗涤,其中乙醇和无水乙醚的体积比为1∶10~20;过滤后干燥,得到三联吡啶双乙酸。
用中性氧化铝色谱柱提纯时使用的洗脱液为石油醚与乙酸乙酯或乙醇的混合液,其中石油醚∶乙酸乙酯或乙醇的体积比为1∶1~1.5。步骤(1)中蒸除溶剂的方法采用减压蒸馏法,其压力为-0.09~-0.1mpa。
应用:利用本发明的三联吡啶双乙酸与zn2+通过溶剂热(甲醇∶dmf=1∶1~1.5,温度100~140℃,聚四氟乙烯内衬的25ml不锈钢反应釜)反应,可以得到五配位结构的zn(ii)配位聚合物,三联吡啶基团近似平行。该锌配合物具有较好的发光性能,荧光激发峰为347nm,发射峰为469nm,相比本发明的三联吡啶双乙酸的荧光光谱(激发峰为339nm,发射峰为450nm)均有明显的红移。本发明的多齿配体三联吡啶双乙酸可作为桥联配体,用于构筑独特结构的zn(ii)配位聚合物,在新型发光材料研究领域具有潜在的应用价值。
图1解析:化学位移的归属如下:12.99(s,2h,-cooh);8.77(d,2h,h6,6’’);8.66(d,4h,h3,3”+h3’,5’);8.03(m,2h,h4,4”);7.80(d,2h,benzene-h);7.53(m,2h,h5,5”);6.73(d,2h,benzene-h);4.21(s,4h,-ch2-)。
图2解析:tpypdca的分子量m=440,质荷比为439.145(-p)的峰可以归属为[m-h]-,质荷比为219.435(-p)的峰可以归属为[m-2h]2-,
图3解析:3424归属为羧基中的o-h的伸缩振动峰,2943归属为烷基(-ch2-)的c-h伸缩振动峰,1599归属为吡啶环中c=n和c=c的伸缩振动峰,1584,1526归属为c=c的面内振动峰,1389归属为o-h的变形振动峰,1214归属为c-n的伸缩振动峰,792归属为苯环中c-h的变形振动峰。
图4解析:三联吡啶双乙酸在丙酮中的最佳激发峰为339nm,荧光发射峰为450nm。
图5解析:三联吡啶双乙酸的锌配合物在丙酮中的最佳激发峰为347nm,荧光发射峰为469nm。
图6解析:本发明的制备步骤图。
图7解析:晶体属斜方晶系,空间群为p-1,r1[i>2σ(i)]=0.064,wr2[i>2σ(i)]=0.1813。晶胞参数:
- 还没有人留言评论。精彩留言会获得点赞!