一种氧导向的7-炔基吲哚类化合物的合成方法与流程
本发明属于7-炔基吲哚类化合物领域,具体涉及一种氧导向的7-炔基吲哚类化合物的合成方法。
背景技术:
吲哚类化合物具有非常重要的生物和药理活性,如抗癌,抗菌,抗高血压等。特殊的化学性质和生物活性使得吲哚类化合物在染料、食品、农业、医药等领域备受关注。因此合成和修饰这类杂环化合物在合成化学中十分重要。炔烃作为材料化学和有机化学中的重要结构基元,是参与多类转化的优越反应前体,采用更有效和更直接的方法合成不同位置取代的炔基吲哚结构受到了广泛的关注。在传统的合成方法中,炔基吲哚衍生物的构建主要由吲哚的c-h卤化以及进一步的交叉偶联反应完成。近年来,伴随着c-h键活化的蓬勃发展,吲哚的直接c-h键官能团化的方法由于简单高效受到了有机化学家的青睐。因此,由过渡金属催化的吲哚未活化的c-h键选择性的直接炔基化反应具有重要的意义。
由于吲哚类化合物具有较多的反应位点,围绕氮原子的位点主要是吲哚的碳2、碳3和碳7位,多反应位点使得对指定位点的选择性c-h活化十分困难。其中,吲哚碳3上位的电子云密度高于其他碳上的电子云密度,这使得3位碳较之其他碳更易于发生金属化作用,从而直接发生c-h炔基化反应(q.yin,h.f.t.klare,m.oestreich,angew.chem.int.ed.,2017,56,3712;h.wang,z.bai,t.jiao,z.deng,h.tong,g.he,q.peng,g.chen,j.am.chem.soc.,2018,140,3542;t.miao,p.li,y.zhang,l.wang,org.lett.,2015,17,832.)。当吲哚底物的碳3位带有位阻基团或者在吲哚的氮原子上面引入一些含氮化合物作为导向基团时,就可以高选择性的合成2-炔基吲哚类化合物(l.yang,l.zhao,c.-j.li,chem.commun.,2010,46,4184;g.l.tolnai,s.ganss,j.p.brand,waserj.org.lett.2013,15,112;z.-z.zhang,b.liu,c.-y.wang,b.-f,shi.org.lett.,2015,17,4094;t.li,z.wang,w.-b.qin,t.-b.wen,chemcatchem.,2016,8,2146;z.ruan,n.sauermann,e.manoni,l.ackermann,angew.chem.int.ed.,2017,129,3220.)。但对于吲哚的碳7位,由于结构本身的电子云密度和位阻的影响,直接的c-h炔基化反应具有极大的挑战。在目前通过c-h活化历程得到7-炔基吲哚衍生物的报道中,均采用吲哚啉作为底物来降低反应的选择性,但该类底物经过碳7位的炔基化反应之后还需通过进一步地被氧化才能得到7-炔基吲哚类化合物(y.wu,y.yang,bingzhou,y.li,j.org.chem.,2015,80,1946;n.jin,c.pan,h.zhang,p.xu,y.cheng,c.zhu,adv.synth.catal.,2015,357,1149.)。这些合成方法虽然在合成方法方面进步很大,但反应步骤较多,反应操作较为繁琐。因此,发展高选择性、步骤经济、绿色高效的7-炔基吲哚类化合物的合成方法是十分必要的。
技术实现要素:
本发明的目的在于针对现有技术的缺点和不足,提供了一种氧导向的7-炔基吲哚类化合物的合成方法。该方法以简单易得的二叔丁基(1h-吲哚-1-基)氧化膦与炔卤为原料,以常见的钯盐作为催化剂,温和的银盐和铜盐作为氧化剂,甲苯为溶剂,采用弱配位的氧作为导向基的策略,选择性地构建了7位炔基化的吲哚衍生物,具有原子经济性高、选择性单一、操作简单安全及底物适用性广等优点,在实际生产和研究中具备良好的应用前景。
本发明的目的通过如下技术方案实现。
一种氧导向的7-炔基吲哚类化合物的合成方法,包含如下步骤:
在反应器中,加入底物二叔丁基(1h-吲哚-1-基)氧化膦类化合物、炔卤,钯盐催化剂、氧化剂、配体和溶剂,在80~100℃下搅拌反应,反应结束后冷却至室温,产物经分离纯化,得到所述7-炔基吲哚类化合物。
进一步地,合成过程的化学反应方程式如下所示:
式中,r1选自氢、4-甲基、4-甲氧基、4-卞氧基、4-氟、4-氯、4-溴、5-甲基、5-甲氧基、5-氟、5-氯和3-甲基中的一种以上;
r2为三异丙基硅基(tips)、叔丁基二甲基硅基(tbdms)中的一种;
x为氯、溴或碘。
进一步地,所述钯盐催化剂为醋酸钯、双(二亚苄基丙酮)钯和三(二亚苄基丙酮)二钯中的一种或两种以上,优选为三(二亚苄基丙酮)二钯。
进一步地,所述钯盐催化剂的加入量与二叔丁基(1h-吲哚-1-基)氧化膦类化合物的摩尔比为0.05~0.15:1,优选为0.1:1。
进一步地,所述炔卤的加入量与二叔丁基(1h-吲哚-1-基)氧化膦类化合物的摩尔比为1.6~2.6:1,优选为1.8:1。
进一步地,所述氧化剂为银盐和铜盐的混合物,由氟化银、碳酸银、氧化铜、钨酸银中的一种以上与三氟甲烷磺酸铜混合得到,优选为碳酸银和三氟甲烷磺酸铜的混合物。
进一步地,所述氧化剂中银盐的加入量与二叔丁基(1h-吲哚-1-基)氧化膦类化合物的摩尔比为1.0~2.0:1,优选为1.8:1;所述氧化剂中铜盐的加入量与二叔丁基(1h-吲哚-1-基)氧化膦类化合物的摩尔比为1.0~2.0:1,优选为1.5:1。
进一步地,所述配体为3-氯吡啶或2-溴吡啶。
进一步地,所述配体的加入量与二叔丁基(1h-吲哚-1-基)氧化膦类化合物的摩尔比为0.1~0.3:1,优选为0.2:1。
进一步地,所述溶剂为甲苯。
进一步地,所述搅拌反应的时间为90℃。
进一步地,所述搅拌反应的时间为6~20小时,优选为8~12小时。
进一步地,所述分离纯化的操作为:将反应液用乙酸乙酯萃取,合并有机相,使用无水硫酸镁干燥,过滤,减压蒸除有机溶剂,得粗产物,经柱层析提纯,得到所述7-炔基吲哚类化合物。
更进一步地,所述柱层析的洗脱液为石油醚和乙酸乙酯按体积比5~50:1的混合溶剂,优选为石油醚和乙酸乙酯按体积比10~30:1的混合溶剂。
本发明合成方法的反应原理是在利用吲哚氮上的二叔丁基氧化膦的位阻效应,使得氧和钯配位后可以选择性地活化吲哚的碳7位,从而形成六元环钯中间体,之后炔卤与其进行氧化加成,再经还原消除,得到7-炔基吲哚类化合物。
与现有技术相比,本发明具有如下优点及有益效果:
(1)本发明发展了二叔丁基(1h-吲哚-1-基)氧化膦与炔卤在氧导向下的交叉偶联反应构建7-炔基吲哚类化合物的合成方法,且其中的基础原料二叔丁基(1h-吲哚-1-基)氧化膦可通过廉价的的二叔丁基氯化膦和吲哚合成,具有原料简单易得、操作安全简单、条件温和、原子经济性高的特点;
(2)本发明合成方法无需二次氧化即可得到目标产物,新颖高效,同时对官能团的容忍性好,因而有望应用于实际工业生产和进一步的衍生化。
附图说明
图1、图2和图3分别是实施例1所得目标产物的氢谱图、碳谱图和磷谱图;
图4、图5和图6分别是实施例2所得目标产物的氢谱图、碳谱图和磷谱图;
图7、图8和图9分别是实施例3所得目标产物的氢谱图、碳谱图和磷谱图;
图10、图11和图12分别是实施例4所得目标产物的氢谱图、碳谱图和磷谱图;
图13、图14、图15和图16分别是实施例5所得目标产物的氢谱图、碳谱图、磷谱图和氟谱图;
图17、图18和图19分别是实施例6所得目标产物的氢谱图、碳谱图和磷谱图;
图20、图21和图22分别是实施例7所得目标产物的氢谱图、碳谱图和磷谱图;
图23、图24和图25分别是实施例8所得目标产物的氢谱图、碳谱图和磷谱图;
图26、图27和图28分别是实施例9所得目标产物的氢谱图、碳谱图和磷谱图;
图29、图30、图31和图32分别是实施例10所得目标产物的氢谱图、碳谱图、磷谱图和氟谱图;
图33、图34和图35分别是实施例11所得目标产物的氢谱图、碳谱图和磷谱图;
图36、图37和图38分别是实施例12所得目标产物的氢谱图、碳谱图和磷谱图;
图39、图40和图41分别是实施例13所得目标产物的氢谱图、碳谱图和磷谱图。
具体实施方式
以下结合具体实施例及附图对本发明的技术方案作进一步详细的描述,但本发明的保护范围及实施方式不限于此。
实施例1
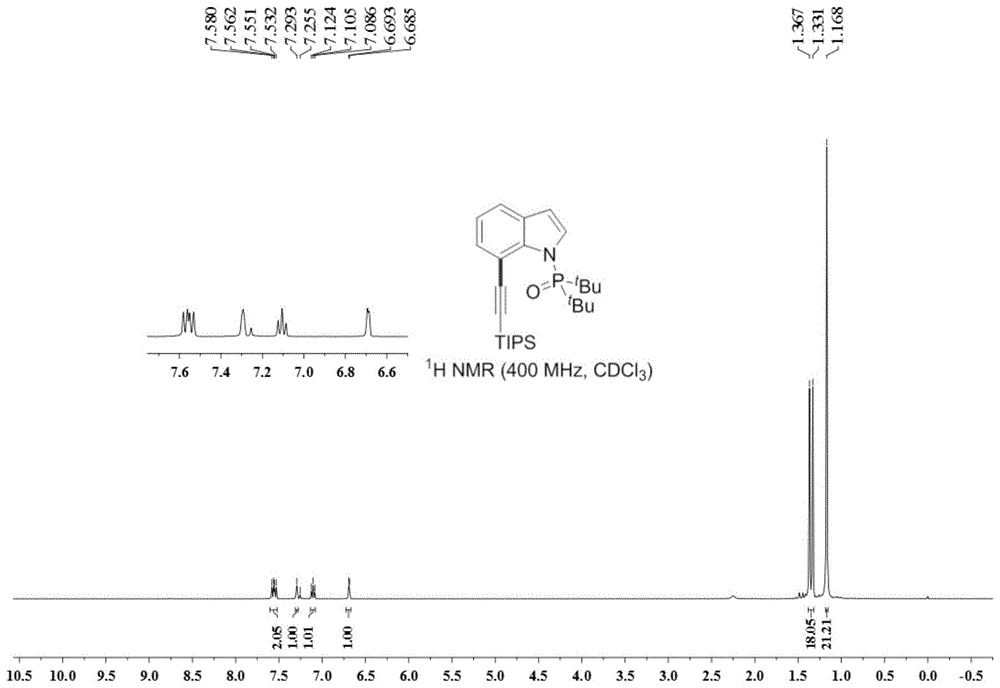
在反应管中加入0.1毫摩尔二叔丁基(1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率81%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图1、图2和图3所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.58-7.53(m,2h),7.29(s,1h),7.11(t,j=7.6hz,1h),6.69(d,j=3.2hz,1h),1.35(d,j=14.4hz,18h),1.17(s,21h);
13cnmr(100mhz,cdcl3)δ=138.3,133.6,131.5(d,j=4.1hz),128.1(d,j=4.8hz),121.6,121.2,112.4,108.0(d,j=5.1hz),107.5,94.8,39.2(d,j=67.3hz),27.1,18.8,11.6;
31pnmr(162mhz,cdcl3)δ=62.51-61.61(m);
ir(kbr)νmax2943,2868,1467,1387,1243,1129,993,879,738,659,474cm-1;
hrms(esi)calcdforc27h45nopsi[m+h]+:458.3003,found458.3008。
经以上数据推断目标产物的结构如下:
实施例2
在反应管中加入0.1毫摩尔二叔丁基(4-甲基-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率82%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图4、图5和图6所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.48(d,j=7.6hz,1h),7.29(s,1h),6.91(d,j=7.2hz,1h),6.71(s,1h),2.50(s,3h),1.35(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=138.0,133.6,131.0(d,j=4.0hz),130.7,127.6(d,j=4.9hz),122.22,109.9,107.8,106.1(d,j=5.1hz),93.8,39.2(d,j=67.3hz),27.1,18.8,11.6;
31pnmr(162mhz,cdcl3)δ=62.17-61.28(m);
ir(kbr)νmax2945,2867,1471,1355,1242,1122,1003,881,864,597,474cm-1;
hrms(esi)calcdforc28h47nopsi[m+h]+:472.3159,found472.3161。
经以上数据推断目标产物的结构如下:
实施例3
在反应管中加入0.1毫摩尔二叔丁基(4-甲氧基-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应6小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率72%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图7、图8和图9所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.48(d,j=7.6hz,1h),7.29(s,1h),6.91(d,j=7.2hz,1h),6.71(s,1h),2.50(s,3h),1.35(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=153.3,139.5,134.9,126.6(d,j=4.5hz),121.7(d,j=4.6hz),107.7,105.5,104.7(d,j=4.9hz),102.2,92.4,55.3,39.2(d,j=67.2hz),27.1,18.8,11.6;
31pnmr(162mhz,cdcl3)δ=62.17-61.28(m);
ir(kbr)νmax2941,2864,1482,1364,1272,1119,881,756,859,476cm-1;
hrms(esi)calcdforc28h47no2psi[m+h]+:488.3108,found488.3111。
经以上数据推断目标产物的结构如下:
实施例4
在反应管中加入0.1毫摩尔(4-(苄氧基)-1h-吲哚-1-基)二叔丁基氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在80℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率82%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图10、图11和图12所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.52-7.45(m,3h),7.38(t,j=7.3hz,2h),7.34-7.30(m,1h),7.20(s,1h),6.88(s,1h),6.63(d,j=8.2hz,1h),5.18(s,2h),1.35(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=152.5,139.6,137.0,134.9,128.5,127.9,127.3,126.6(d,j=4.7hz),122.1(d,j=4.5hz),107.6,105.7,104.9(d,j=5.1hz),103.6,92.6,70.0,39.2(d,j=67.2hz),27.1,18.8,11.6;
31pnmr(162mhz,cdcl3)δ=62.56-61.66(m);
ir(kbr)νmax2940,2864,1481,1366,1275,1117,1018,881,747,657,473cm-1;
hrms(esi)calcdforc34h51no2psi[m+h]+:564.3421,found564.3429。
经以上数据推断目标产物的结构如下:
实施例5
在反应管中加入0.1毫摩尔二叔丁基(4-氟-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在100℃、转速700rpm下搅拌反应20小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率79%。
所得目标产物的氢谱图、碳谱图、磷谱图和氟谱图分别如图13、图14、图15和图16所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.53-7.49(m,1h),7.27(s,1h),6.83-6.79(m,2h),1.36(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=156.1(d,j=248.8hz),140.5(d,j=9.4hz),134.4(d,j=7.4hz),128.0(d,j=4.6hz),120.4(dd,j=22.2,4.5hz),108.7(d,j=4.0hz),107.1(d,j=18.8hz),106.7,103.3(d,j=5.0hz),94.2,39.3(d,j=66.8hz),27.1,18.8,11.5;
31pnmr(162mhz,cdcl3)δ=63.40-62.48(m);
19fnmr(376mhz,cdcl3)δ=-120.43(t,j=7.0hz);
ir(kbr)νmax2946,2871,1593,1479,1361,1247,1116,1007,882,799,662,480cm-1;
hrms(esi)calcdforc27h44fnopsi[m+h]+:476.2908,found476.2910。
经以上数据推断目标产物的结构如下:
实施例6
在反应管中加入0.1毫摩尔二叔丁基(4-氯-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔2-溴吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率60%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图17、图18和图19所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.49(d,j=8.0hz,1h),7.34(s,1h),7.12(d,j=8.0hz,1h),6.84(d,j=2.0hz,1h),1.35(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=138.8,133.9,130.2(d,j=4.1hz),128.7(d,j=4.5hz),126.3,121.5,111.2,106.6,106.2(d,j=4.8hz),95.9,39.3(d,j=66.6hz),27.1,18.8,11.5;
31pnmr(162mhz,cdcl3)δ=63.49-62.60(m);
ir(kbr)νmax2946,2868,1468,1348,1239,1122,998,884,812,751,637,472cm-1;
hrms(esi)calcdforc27h43clnnaopsi[m+na]+:514.2432,found514.2438。
经以上数据推断目标产物的结构如下:
实施例7
在反应管中加入0.1毫摩尔二叔丁基(4-溴-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率53%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图20、图21和图22所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.42(d,j=8.2hz,1h),7.34(s,1h),7.28(d,j=8.0hz,1h),6.80(d,j=2.0hz,1h),1.35(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=138.3,134.0,132.0(d,j=3.9hz),128.6(d,j=4.6hz),124.7,115.0,111.7,108.1(d,j=4.6hz),106.6,96.1,39.3(d,j=66.6hz),27.1,18.8,11.5;
31pnmr(162mhz,cdcl3)δ=63.60-62.70(m);
ir(kbr)νmax2942,2866,1466,1342,1238,1123,993,878,811,744,657,473cm-1;
hrms(esi)calcdforc27h43brnnaopsi[m+na]+:558.1927,found558.1931。
经以上数据推断目标产物的结构如下:
实施例8
在反应管中加入0.1毫摩尔二叔丁基(5-甲基-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率79%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图23、图24和图25所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.37(s,1h),7.33(s,1h),7.25(s,1h),6.61(d,j=2.0hz,1h),2.38(s,3h),1.34(d,j=14.4hz,18h),1.17(s,21h);
13cnmr(100mhz,cdcl3)δ=136.8,134.6,131.8(d,j=4.2hz),130.9,128.2(d,j=4.9hz),121.3,111.9,107.6(d,j=5.7hz),94.2,39.2(d,j=67.5hz),27.1,20.7,18.8,11.6;
31pnmr(162mhz,cdcl3)δ=61.91-61.02(m);
ir(kbr)νmax2944,2867,1466,1381,1239,1124,991,878,753,661,608,473cm-1;
hrms(esi)calcdforc28h47nopsi[m+h]+:472.3159,found472.3163。
经以上数据推断目标产物的结构如下:
实施例9
在反应管中加入0.1毫摩尔二叔丁基(5-甲氧基-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.10毫摩尔碳酸银、0.20毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比10:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率48%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图26、图27和图28所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.27(d,j=2.4hz,1h),7.19(d,j=2.4hz,1h),7.03(d,j=2.4hz,1h),6.61(d,j=3.2hz,1h),3.81(s,3h),1.33(d,j=14.4hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=154.4,133.5,132.4(d,j=4.2hz),128.8,121.8,113.0,107.8(dd,j=4.4,3.3hz),107.0,104.0(d,j=1.4hz),94.8,39.2(d,j=67.3hz),27.1,18.8,11.5;
ir(kbr)νmax2045,2870,1679,1595,1467,1387,1228,1947,805,660,472cm-1;
31pnmr(162mhz,cdcl3)δ=62.12-61.21(m);
hrms(esi)calcdforc28h47no2psi[m+h]+:488.3108,found488.3111。
经以上数据推断目标产物的结构如下:
实施例10
在反应管中加入0.1毫摩尔二叔丁基(5-氟-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔2-溴吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率56%。
所得目标产物的氢谱图、碳谱图、磷谱图和氟谱分别如图29、图30、图31和图32所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.34(s,1h),7.28(d,j=9.6hz,1h),7.20(d,j=8.0hz,1h),6.65(s,1h),1.34(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=157.7(d,j=237.4hz),135.1,132.4(dd,j=10.3,4.1hz),129.7(d,j=4.6hz),120.4(d,j=25.6hz),113.3(d,j=10.2hz),107.9(t,j=4.6hz),106.7(d,j=22.9hz),106.2(d,j=2.0hz),96.3,39.3(d,j=67.0hz),27.1,18.8,11.5;
31pnmr(162mhz,cdcl3)δ62.90-62.00(m);
19fnmr(376mhz,cdcl3)δ-123.43(t,j=9.0hz);
ir(kbr)νmax2945,2870,1581,1467,1381,1234,1138,985,871,802,662,476cm-1;
hrms(esi)calcdforc27h44fnopsi[m+h]+:476.2908,found476.2913。
经以上数据推断目标产物的结构如下:
实施例11
在反应管中加入0.1毫摩尔二叔丁基(5-氯-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔2-溴吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率46%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图33、图34和图35所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.49(d,j=8.0hz,1h),7.34(s,1h),7.12(d,j=8.0hz,1h),6.84(d,j=2.0hz,1h),1.35(d,j=14.8hz,17h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=138.8,133.9,130.2(d,j=4.1hz),128.7(d,j=4.5hz),126.3,121.5,111.2,106.6,106.2(d,j=4.8hz),95.9,39.3(d,j=66.6hz),27.1,18.8,11.5;
31pnmr(162mhz,cdcl3)δ63.49-62.60(m);
ir(kbr)νmax2939,2868,1459,1372,1235,1132,997,879,757,671,474cm-1;
hrms(esi)calcdforc27h43clnnaopsi[m+na]+:514.2432,found514.2435。
经以上数据推断目标产物的结构如下:
实施例12
在反应管中加入0.1毫摩尔二叔丁基(3-甲基-1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.20毫摩尔碳酸银、0.10毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔三异丙基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率84%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图36、图37和图38所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.57(d,j=7.2hz,1h),7.46(d,j=7.6hz,1h),7.12(t,j=7.2hz,1h),7.04(s,1h),2.29(s,3h),1.34(d,j=14.8hz,18h),1.16(s,21h);
13cnmr(100mhz,cdcl3)δ=138.9,133.5,132.3(d,j=3.9hz),125.2(d,j=2.5hz),121.1,119.1,116.6(d,j=5.2hz),112.4,107.6,94.6,39.1(d,j=67.7hz),27.1,18.8,11.5,9.7;
31pnmr(162mhz,cdcl3)δ61.79-61.08(m);
ir(kbr)νmax2943,2869,1466,1390,1228,1128,1005,908,743,660,474cm-1;
hrms(esi)calcdforc28h47nopsi[m+h]+:472.3159,found472.3161。
经以上数据推断目标产物的结构如下:
实施例13
在反应管中加入0.1毫摩尔二叔丁基(1h-吲哚-1-基)氧化膦、0.01毫摩尔三(二亚苄基丙酮)二钯、0.18毫摩尔碳酸银、0.15毫摩尔三氟甲烷磺酸铜、0.02毫摩尔3-氯吡啶、0.18毫摩尔叔丁基二甲基硅基炔溴,1.5毫升甲苯作为溶剂,在90℃、转速700rpm下搅拌反应12小时后,停止加热及搅拌,冷却至室温,加入5ml水,用乙酸乙酯萃取3次,合并有机相并使用0.5g无水硫酸镁干燥,过滤,减压浓缩,再通过柱层析分离纯化,所用的柱层析洗脱液为体积比30:1的石油醚:乙酸乙酯混合溶剂,得到目标产物,产率84%。
所得目标产物的氢谱图、碳谱图和磷谱图分别如图39、图40和图41所示,结构表征数据如下所示:
1hnmr(400mhz,cdcl3)δ=7.54(d,j=7.6hz,1h),7.30(d,j=1.6hz,1h),7.11(t,j=7.6hz,1h),6.69(s,1h),1.35(d,j=14.8hz,18h),0.99(s,9h),0.21(s,6h);
13cnmr(100mhz,cdcl3)δ=138.7,133.0,131.5(d,j=4.0hz),128.2(d,j=4.8hz),121.6,121.3,112.1,108.1(d,j=5.0hz),106.1,96.2,39.3(d,j=67.3hz),27.1,26.4,17.0,-4.5;
31pnmr(162mhz,cdcl3)δ62.80-61.92(m);
ir(kbr)νmax2951,1686,1490,1399,1257,1124,988,818,739,661,598,525cm-1;
hrms(esi)calcdforc24h39nopsi[m+h]+:416.2533,found416.2531。
经以上数据推断目标产物的结构如下:
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!