一种I型N-聚糖天线的合成方法与流程
本发明属于医药技术领域,具体而言,涉及一种i型n-聚糖天线的合成方法。
背景技术:
在糖脂和糖蛋白中广泛存在n-聚糖(天冬酰胺连接的聚糖)天线。从结构上讲,天线可分为i型和ii型两类,i型天线含有α-2,6连接的唾液残基,ii型天线含有α-2,3连接的唾液残基。鉴于n-聚糖在维持糖蛋白正常功能和病毒宿主识别中的关键作用,具有明确化学结构的n-聚糖的化学合成已成为糖化学中一个广泛研究的领域。
5位ac保护的唾液酸
尽管n-聚糖具有广泛的应用前景促使人们对其研究不断深入,而n-聚糖天线又是合成n-聚糖的核心也是合成的难点。首先唾液酸α糖苷键的构建是一个巨大的挑战,而后续的高效率糖链的延伸也是个不小的挑战。之前有不少科学家对其进行合成,但合成的效率及其灵活性却不尽人意。
2009年samuelj.danishefsky小组合成含有i型n-聚糖天线的单聚体igg。该组在合成采用的是从后向前的链式合成,但其每一步都需要将受体进行保护基操作,在裸露受体羟基时需要用到水合肼等剧毒物,在脱出ca时的产率只有52%。
2014年shang-chenghung小组利用一锅法来合成3糖i型n-聚糖天线。但其合成3糖的产率45%且灵活性差,在进行末端唾液酸的延伸时需要用剧烈的条件先脱除phth保护基,若需要延长糖链其糖异头位ome基的脱除及制成给体也有很大的限制。
2016年koichifukase小组合成在合成n-聚糖时,对其天线进行2+2的合成方式。在含唾液酸2糖模块的构建时,α选择性只有16:1,产率也只有85%,在将该2糖模块改装成给体时产率操作繁琐产率偏低且需要用到昂贵的ir(cod)(pph2me)2]pf6催化,其合成后的衍生化也有很大的困难。
因此对i型n-聚糖天线的高效率和灵活性还有很大的困难。迫切需要建立一条有效的天线组装路线,以促进n-聚糖的合成,并进行之后的结构-活性关系的研究。
技术实现要素:
本发明要解决的技术问题是需要建立一条有效的i型n-聚糖天线组装路线,以促进n-聚糖的合成以及之后的结构-活性关系的研究。
为解决上述技术问题,本发明采取的技术方案是:一种i型n-聚糖天线的合成方法,包括以下步骤:
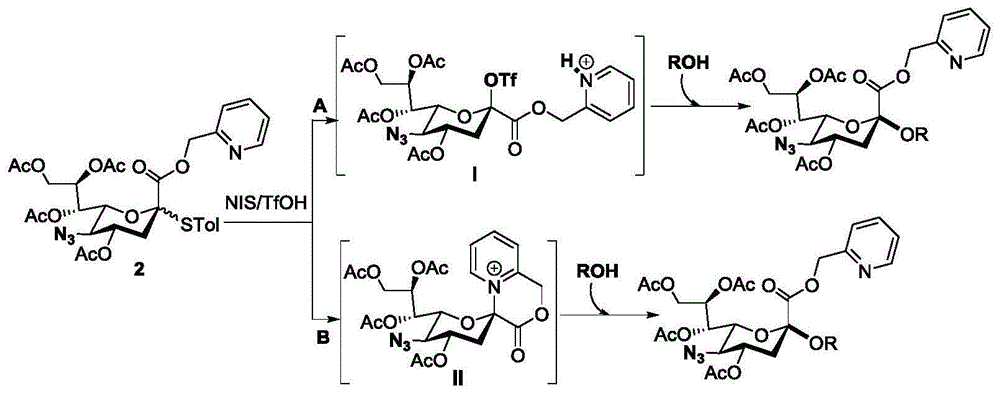
(1)将糖基给体化合物1和糖基受体化合物2溶于第一溶剂中,加入干燥剂,加入nis和tfoh,发生糖苷化反应,得到化合物3,所述糖基给体化合物1、糖基受体化合物2、nis和tfoh的摩尔比为1:1:1:1~5:1:5:5,所述化合物1在第一溶剂中的浓度为0.001~1mol/l,所述第一溶剂为二氯甲烷;所述糖苷化反应温度为-40℃~0℃;
(2)将化合物3溶于第二溶剂中,在45~50℃下加入醋酸铜反应得到化合物4,所述第二溶剂为甲醇,所述化合物3与醋酸铜的摩尔比为1:1~1:10,所述化合物3在第二溶剂中的浓度为0.001~1mol/l;
(3)化合物4溶于第三溶剂中,在室温下加入硫代乙酸反应得到化合物5,所述化合物4与硫代乙酸的摩尔比为1:10~1:100,所述化合物4在第三溶剂中的浓度为0.001~1mol/l;
(4)将化合物5、ph3p、pd(pph3)2cl2、cui溶解在第四溶剂,降温到-78℃~-40℃后,加入对甲氧基苯乙炔在第五溶剂中的溶液,升温至50~100℃后,反应得到化合物6,所述化合物5、ph3p、pd(pph3)2cl2、cui和对甲氧基苯乙炔的摩尔比为1:0.2:0.1:0.2:1.2~1:0.5:0.2:0.5:1.6,化合物5在第四溶剂中的浓度为0.01~1mol/l;
(5)将化合物6和糖基受体化合物7溶于第六溶剂中,并加入干燥剂,在-50℃~0℃,路易斯酸和nis催化下发生糖苷化反应得到化合物8;
(6)将化合物8、ph3p、pd(pph3)2cl2、cui、溶解在第七溶剂中,降温到-78℃~-40℃后,加入对甲氧基苯乙炔在第八溶剂中的溶液,升温至50~100℃后,反应得到化合物9;所述化合物8、ph3p、pd(pph3)2cl2、cui和对甲氧基苯乙炔的摩尔比为1:0.2:0.1:0.2:1.2~1:0.5:0.2:0.5:1.6,化合物8在第七溶剂中的浓度为0.01~1mol/l;
(7)将化合物9和糖基受体化合物10溶于第九溶剂中,并加入干燥剂,在-50℃~0℃,催化剂作用下反应得到化合物11。
步骤(1)中,所述糖苷化反应温度优选的为-40℃~-20℃;所述化合物1在第一溶剂中的浓度优选的为0.001~0.003mol/l,所述干燥剂选自分子筛,优选的为
tfoh的用量对1和2的糖苷化反应有很大影响。当以催化量(0.2eq)使用tfoh时,仅分离出9%的糖苷化产物3。随着tfoh用量从0.5eq增加到1.0eq,糖苷化产物3的收率从34%逐渐上升到90%。令人惊讶的是,使用当量的tfoh,也能保持良好的唾液酸化立体选择性(α/β=25:1)。再加大tfoh的量到5个当量产率和比例也没有变化。为了评估硫唾液酸供体的非对映异构体对唾液酸化结果的影响,供体1α也在与3相同的条件下糖苷化。虽然观察到产量和立体选择性略有下降,但它们仍处于良好水平(83%,α/β=20:1)(表1)。
表1糖苷化条件优化
步骤(2)中,所述化合物3与醋酸铜的摩尔比优选为1:1~1:4,所述化合物3在第二溶剂中的浓度优选为0.02~0.004mol/l。
所述第二溶剂为选自甲醇,dcm或丙酮中的一种或多种,优选为甲醇。
当所述第二溶剂为体积比1:1的dcm和meoh的混合溶剂时,步骤2反应产率为60%,当所述第二溶剂为为体积比1:1的丙酮和甲醇的混合溶剂时,步骤2反应产率为45%。当以甲醇为反应溶剂时,步骤2反应产率为90%。
步骤(3)中,所述第三溶剂为吡啶;所述化合物4与硫代乙酸的摩尔比优选为1:10~1:30,所述化合物4在第三溶剂中的浓度优选为0.001~0.6mol/l。
我们首先尝试用二氯甲烷和三乙胺混合溶剂,与硫代乙酸对化合物4进行还原乙酰化,但反应没能成功,我们换吡啶和二氯甲烷为溶剂与硫代乙酸进行还原氨化以一般产率拿到我们所需要的产物5。最后我们直接以吡啶为溶剂与硫代乙酸进行还原氨化以72%的产率得到所需产物5。三乙胺和吡啶都用于中和体系里的酸增强硫代乙酸的亲核性。
步骤(4)中,所述第四和第五有机溶剂相同或不同的选自n,n-二甲基甲酰胺(dmf)、二异丙胺(i-pr2nh)、四氢呋喃(thf)、二氯甲烷(dcm)、丙酮、甲醇(meoh)或乙醇(etoh)中的一种或多种,优选的,所述第四有机溶剂为dmf和i-pr2nh的混合溶液,更优选的,所述dmf和i-pr2nh的体积比为1:3~5,能够更好的溶解化合物5;优选的,所述第五有机溶剂为dmf。所述化合物5、ph3p、pd(pph3)2cl2、cui和对甲氧基苯乙炔的摩尔比优选的为1:0.2:0.1:0.2:1.2~1:0.45:0.1:0.45:1.5;化合物5在第四有机溶剂中的浓度优选为0.01~0.03mol/l。
步骤(5)中,所述第六有机溶剂为甲苯、二氯甲烷、乙醚、丙酮、thf中的一种或多种,优选的为二氯甲烷。所述的路易斯酸为tmsotf,所述化合物7和路易斯酸、nis的摩尔比为1:0.1:1~1:1:2;优选的为1:0.3:1.2~1:0.5:1.5;化合物6和化合物7的摩尔比为1:1~1:5,优选的为1:1~1:1.2;式i所示的糖苷化给体与第六有机溶剂的质量体积比为20~100mg/ml,优选为20~50mg/ml;所述干燥剂选自分子筛,优选的为
步骤(6)中,所述第七和第八有机溶剂相同或不同的选自n,n-二甲基甲酰胺(dmf)、二异丙基胺(i-pr2nh)、四氢呋喃(thf)、二氯甲烷(dcm)、丙酮、甲醇(meoh)或乙醇(etoh)中的一种或多种。优选的,所述第七有机溶剂为dmf和ipr2nh的混合溶液,更优选的,所述dmf和i-pr2nh的体积比为1:3~1:5,能够更好的溶解化合物8;优选的,所述第八有机溶剂为dmf。所述化合物ii、ph3p、pd(pph3)2cl2、cui和对甲氧基苯乙炔的摩尔比为1:0.2:0.1:0.2:1.2~1:0.5:0.2:0.5:1.6;优选的为1:0.2:0.1:0.2:1.2~1:0.45:0.1:0.45:1.5;化合物8在第七有机溶剂中的浓度为0.01~1mol/l,优选为0.01~0.03mol/l。
步骤(7)中,所述第九有机溶剂为甲苯、二氯甲烷、乙醚、丙酮、thf中的一种或多种,优选的,为二氯甲烷;所述的路易斯酸为tmsotf,所述化合物9和路易斯酸、nis的摩尔比为1:0.1:1~1:1:2;优选的为1:0.3:1.2~1:0.5:1.5;化合物6和化合物7的摩尔比为1:1~1:5,优选的为1:1~1:1.2;式i所示的糖苷化给体与第六有机溶剂的质量体积比为20~100mg/ml,优选为20~50mg/ml;所述干燥剂选自分子筛,优选的为
优选的,步骤(1)~(7)中的任一步骤在惰性气体保护条件下进行,所述惰性气体选自氮气、氩气或氦气。
i型天线11的合成起始于1和一级羟基受体2之间的耦合,受体2在的异头位保护用ip。在nis/tfoh的作用下,在以ch2cl2为溶剂发生糖苷化反应,以高立体选择性、高效地得到了二糖唾液苷3(90%和α-异构体)。在cu(oac)2的作用下,3的参与基团去除和甲基酯化反应均得到了较好的效果,使反应顺利进行得到4(90%)。硫代乙酸/吡啶将4中的n3转化为相应的乙酰胺基得到潜在双糖供体化合物5,分离率高达72%,然后通过sonogashira反应进行活化,得到双糖mpep供体化合物6(87%)。在标准活化条件下,供体6与7有效地糖基化,得到三糖潜在供体化合物8,然后通过sonogashira反应(83%)转化为三糖mpep供体9。随后的聚糖链延伸需要三糖mpep供体和10的惰性直立键oh。令人高兴的是,9和10之间的糖苷化没有观察到糖基化效率的降低,并且四糖潜在供体11获得了良好的82%的产量,这是为i型天线安装打好基础。因此,通过结合2-糖苷化方法和mpep糖基化的方案,i型天线11可以通过最长7步线线性合成从容易获得的单糖切块获得,总收率为29%。
pic基团发挥其手性控制作用的可能机理如下:第一种是pic促进的三氟甲磺酸化唾液形成机制(图1,机制a),另一种是pic参与机制(图1,机制b)。在第一种机制中,使用的当量三氟甲磺酸使pic基团的氮原子质子化,从而导致形成强烈吸电子吡啶甲基盐,与c5位的n3吸电子作用结合,使在nis/tfoh作用下产生不稳定的一氧鎓,从而促进三氟甲磺酸根阴离子的β-攻击。三氟甲磺酸化得唾液酸中间物i。然后三氟甲磺酸化唾液酸中间物与受体进行sn2取代,得到α-唾液酸衍生物。在第二种机制中,即使在化学计量量的三氟甲磺酸存在下,pic基团在c5-n3的协助下直接作为一个参与基团,形成热力学更稳定得6元中间体ii,通过该中间体,受体随后发生sn2进攻也得到α-选择性糖苷化产物。
有益效果:
1)本发明解决了唾液酸糖苷化立体选择性差和产率低的问题。
2)本发明高效高立体选择性的制备了i型n-聚糖天线。将大大推进n-聚糖类化合物的活性机理研究及其结构-活性关系的研究。
3)本发明利用可靠的唾液酸糖苷化方法和潜在活性”糖苷合成策略为特征的mpep糖基化方案。大大缩短了合成路线,为大量合成提供了可能。
附图说明
图1为糖苷化高立体选择性的机理推测。
具体实施方式
根据下述实施例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施例所描述的内容仅用于说明本发明,而不应当也不会限制权利要求书中所详细描述的本发明。
本发明中,缩写代表含义如下:
pic2-吡啶甲基(2-picolyl),结构式:
nisn-碘代丁二酰亚胺(n-iodosuccinimide)
ip:
mpep
phth邻苯二甲酰基(phthaloyl)
tfoh三氟甲磺酸(trifluoromethanesulfonicacid)
tmsotf三氟甲磺酸三甲基硅酯(trimethylsilyltrifluoromethanesulfonate)
bz苯甲酰基(benzoyl)
bn苄基(benzyl)
ac乙酰基(acetyl)
以下每步反应得到的产物纯度较高,在400兆核磁共振氢谱中见不到杂质峰。
本发明中未提及的原料均为市售。
反应原料的合成:
1、适当基团保护的糖1-1、2-1、7-1、10-1按照本领域常规方法合成。
2、化合物1的合成
化合物1的合成参考文献;yu,c.-s.;niikura,k.;lin,c.-c.;wong,c.-h.angew.chem.int.ed.2001,40,2900-2903.具体合成步骤如下:
将化合物1-1(100mg,0.17mmol)溶解在吡啶(4.0ml)中,常温下加入licl(22mg,0.52mmol).将此反应体系加热回流120℃下反应24小时.减压浓缩除去吡啶,将剩余物溶于二氯甲烷中,并依次用1mol/lhcl、饱和nacl洗,无水硫酸钠干燥,抽滤,减压浓缩得粗产物,不需要进一步纯化。将上述得到的化合物溶于干燥的dmf(2ml),室温加入picbr·hbr(90mg,0.36mmol)和k2co3(95mg,0.69mmol)在n2保护下,体系室温下搅拌5小时,tlc跟踪显示原料反应完全,将反应体系用二氯甲烷萃取,并依次用1mol/lhcl、饱和碳酸氢钠、饱和nacl洗,无水硫酸钠干燥。过滤减压浓缩得粗产物,最后再柱层析(pe:ea:dcm=3:1:1)得到白色固体化合物1(90.6mg,80%for2steps)(α/β=1:3).2α:[α]d25=-22.5(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ8.58(d,j=4.0hz,1h),7.73(td,j=1.6,7.6hz,1h),7.38(t,j=8.4hz,3h),7.24(dd,j=5.2,8.0hz,1h),7.13(d,j=7.6hz,2h),5.51(dd,j=1.2,8.4hz,1h),5.27-5.24(m,1h),5.24(d,j=14.0hz,1h),5.11(d,j=13.6hz,1h),5.06(ddd,j=4.8,9.6,11.6hz,1h),4.38(dd,j=2.4,12.4hz,1h),4.32(dd,j=4.0,12.8hz,1h),3.86(dd,j=1.6,10.4hz,1h),3.21(t,j=10.4hz,1h),3.05(dd,j=4.8,13.2hz,1h),2.35(s,3h),2.20(s,3h),2.10(s,3h),2.05(s,3h),1.91(s,3h),1.89(t,j=12.4hz,1h);13cnmr(100mhz,cdcl3)δ170.8,170.0,169.8,169.6,166.9,154.8,149.4,140.3,137.0,136.4,129.8,125.0,123.0,121.3,86.8,73.4,71.7,69.1,68.2,67.6,61.7,60.2,37.7,21.4,21.0,20.9,20.8;hrms(esi)calcdforc30h34n4o11sna[m+na]+681.1837,found681.1848.2β:[α]d25=-80(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ8.58-8.56(m,1h),7.72(td,j=1.6,7.6hz,1h),7.30-7.23(m,4h),7.09(d,j=7.6hz,2h),5.63(dd,j=2.0,5.6hz,1h),5.48(ddd,j=4.8,9.6,11.6hz,1h),5.21(d,j=13.6hz,1h),5.16-5.13(m,1h),5.14(d,j=13.2hz,1h),4.39(dd,j=2.0,10.4hz,1h),4.38(dd,j=2.4,12.4hz,1h),4.26(dd,j=6.0,12.4hz,1h),3.34(t,j=10.0hz,1h),2.80(dd,j=4.8,14.0hz,1h),2.29(s,3h),2.16(s,3h),2.11(s,3h),2.08(s,3h),2.021(s,3h),2.015(dd,j=11.6,14.0hz,1h);13cnmr(100mhz,cdcl3)δ170.4,170.1,169.8,169.6,166.9,155.0,149.2,140.2,136.9,136.0,130.1,125.2,123.0,121.7,88.2,71.6,70.8(2c),69.2,67.7,61.7,60.3,36.5,21.3,21.0,20.9,20.8,20.7;hrms(esi)calcdforc30h35n4o11s[m+h]+659.2018,found659.2049.
3、化合物2的合成方法
先将邻碘苯酚(803mg,3.65mmol)溶解在chcl3(7ml)中,加入称量好的tbab(522mg,1.6mmol)和k2co3(15ml,0.25mmol/l)40℃.搅拌10分钟后加入全乙酰基保护半乳糖溴苷,2-1(1.0g,2.4mmol)的chcl3(7ml)溶液.在同样的温度下搅拌过夜,点板监控反应完全,冷却到室温加入大量的dcm。首先用水洗两次,再用饱和nahco3洗两次,合并有机相用无水na2so4干燥,过滤旋干滤液过层析柱用(pe:ea=5:1)得到白色固体2-2(937mg,70%)[α]d25=-32.0(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ7.80(dd,j=1.6,7.6hz,1h),7.32-7.28(m,1h),7.10(dd,j=1.2,8.0hz,1h),6.85(td,j=1.6,7.6hz,1h),5.65(dd,j=7.6,10.0hz,1h),5.48(dd,j=1.2,3.6hz,1h),5.13(dd,j=3.2,10.4hz,1h),5.04(d,8.0hz,1h),4.29(dd,j=6.8,11.2hz,1h),4.19(dd,j=6.0,11.2hz,1h),4.12-4.08(m,1h),2.20(s,3h),2.12(s,3h),2.08(s,3h),2.03(s,3h);13cnmr(100mhz,cdcl3)δ170.4(2c),170.3,169.4,156.1,139.8,129.6,125.0,116.0,100.3,86.9,71.3,71.0,68.2,66.9,61.5,21.5,20.8(2c),20.7;hrms(esi)calcdforc21h24io12[m+hco2]-595.0307,found595.0292.
将化合物2-2(800mg,1.45mmol)溶于无水甲醇(6ml)中,加入甲醇钠(16mg,0.29mmol)在室温和n2保护下.反应体系在这个温度下搅拌1h,点板监控反应完全,加入甲醇稀释,用酸性树脂调节ph在7左右.过滤、减压旋干得粗产物不需要进一步纯化。将上述粗产物溶于乙腈(15ml)中,加入csa(67mg,0.29mmol)和phch(ome)2(0.435ml,2.9mmol)在室温和n2保护下.反应体系升温至60℃反应过夜。点板监控反应完全,将体系冷却至室温,加入et3n淬灭反应,倒入t冰水和石油醚的混合体系中(v/v=1:1)搅拌.过滤得到白色固体,再用小极性溶剂冲洗,烘干得到白色固体化合物2-3(571.2mg,84%):[α]d25=-23.7(c0.3,chcl3);1hnmr(400mhz,cdcl3)δ7.78(dd,j=1.6,8.0hz,1h),7.55-7.53(m,2h),7.41-7.38(m,3h),7.32(td,j=1.6,8.4hz,1h),7.22(dd,j=1.6,8.4hz,1h),6.85(td,j=1.6,7.6hz,1h),5.61(s,1h),4.84(d,j=8.0hz,1h),4.44(dd,j=1.6,12.8hz,1h),4.32(d,j=4.0hz,1h),4.18-4.14(m,2h),3.85(dd,j=4.0,10.0hz,1h),3.67(brs,1h),13cnmr(100mhz,cdcl3)δ156.2,139.3,137.4,129.8,129.5,128.4,126.6,125.1,117.0,103.3,101.7,88.0,75.1,72.3,71.7,69.2,67.2;hrms(esi)calcdforc19h19io6na[m+na]+493.0119,found493.0114.
将化合物2-3(280mg,0.6mmol)和二丁基氧化硒(149.6mg,0.6mmol)溶于干燥得甲苯中,在室温和n2保护下,加热至回流,在此温度下反应3h.冷却至室温,减压条件下移去溶剂得粗产物。将上述粗产物溶于干燥得dmf(3ml),加入csf(93mg,0.61mmol)和bnbr(80μl,0.67mmol)在室温和n2保护下.反应体系加热至80℃,在这个温度下搅拌过夜.将反应体系冷却至室温,加入二氯甲烷稀释.the依次用水、饱和nacl洗,最后用无水na2so4干燥.过滤、减压旋干得到粗产物,不需要进一步纯化。将上述粗产物溶于吡啶4ml,加入苯甲酰氯(0.5ml)常温下,反应体系在这个温度下搅拌1h,点板监控反应完全,加入甲醇淬灭反应,依次用水,1nhcl、饱和nahco3、饱和nacl洗、用无水na2so4干燥,过滤减压旋干,过层析柱得到白色固体2-4(250mg,63%for3steps):[α]d25=+13.9(c0.75,chcl3);1hnmr(400mhz,cdcl3)δ8.04(d,j=7.6hz,2h),7.65(d,j=8.0hz,1h),7.59(dd,j=7.2,16.4hz,3h),7.43(t,j=7.6hz,2h),7.37-7.32(m,4h),7.24-7.16(m,6h),6.73(t,j=7.2hz,1h),6.02(t,j=9.2hz,1h),5.53(s,1h),5.08(d,j=8.0hz,1h),4.72(ab,2h),4.36(d,j=12.4hz,1h),4.31(d,j=3.2hz,1h),4.12(d,j=12.4hz,1h),3.83(dd,j=3.2,10.4hz,1h),3.54(brs,1h);13cnmr(100mhz,cdcl3)δ165.2,156.4,139.6,137.8,137.6,133.0,130.6,130.1,129.3,129.1,128.4,128.3(2c),127.9,127.8,126.6,124.7,117.0,101.3,100.8,87.5,77.0,72.9,71.1,70.0,69.1,67.1;hrms(esi)calcdforc33h29io7na[m+na]+687.0850,found687.0862.
将化合物2-4(285mg,0.429mmol)溶于干燥thf(3.0ml)加入1nbh3·thf(2.15ml,2.15mmol)和cu(otf)2(31.2mg,0.086mmol)冰浴下,n2保护下。再移至室温反应3小时,tlc监控反应完全,加入et3n淬灭反应.用ch2cl2稀释,依次用水、饱和nacl溶液洗涤,无水硫酸钠干燥,过滤、减压浓缩浓缩得粗产物,最后再柱层析(pe:ea=5:10得到白色固体化合物2(237.3mg,83%):[α]d25=+122.4(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ7.43-7.29(m,10h),7.03(d,j=8.8hz,2h),6.85(d,j=9.2hz,2h),5.45(d,j=3.6hz,1h),4.99(t,j=10.8hz,2h),4.92(d,j=10.8hz,1h),4.73(d,j=11.2hz,1h),4.25(dd,j=8.8,10.4hz,1h),3.92(dt,j=2.8,10.0hz,1h),3.80-3.72(m,3h),3.77(s,3h),3.47(dd,j=3.6,10.4hz,1h);13cnmr(100mhz,cdcl3)δ155.6,150.4,137.9(2c),128.7(2c),128.2(2c),128.1(2c),118.3,114.8,97.8,80.1,77.7,75.7,75.3,72.2,63.5,61.5,55.8;hrms(esi)calcdforc27h29n3o6na[m+na]+514.1949,found514.1957.
4、化合物7的合成参考文献:hu,y.;yu,k.;shi,l.-l.;liu,l.;sui,j.-j.;liu,d.-y.;xiong,b.;sun,j.-s.j.am.chem.soc.2017,139,12736-12744.具体实验步骤如下:
将化合物7-1(1.1g,1.7mmol)溶解于无水甲醇(10ml),加入naome(9.18mg,0.17mmol)在室温和氮气保护下。在这个温度下搅拌1h,用酸性树脂条件体系ph至7左右。过滤、减压旋去滤液得粗产物,不需要进一步纯化。将上述粗产物溶于dmf(8ml)中,加入phch(ome)2(0.56ml,3.68mmol)和tsoh(56mg,0.29mmol)在室温和氮气保护下。体系在此温度下搅拌过夜,点板监控反应完全,用三乙胺淬灭反应,用二氯甲烷稀释,依次用饱和nahco3,饱和nacl洗,再用无水na2so4干燥。过滤、减压旋去滤液,过层析柱(peea=3:1)得白色固体化合物7-2(908.9mg,88%for2steps):[α]d25=+42.3(c1.0,chcl3);1hnmr(400mhz,acetone-d6)δ7.86(s,4h),7.67(dd,j=7.8,1.5hz,1h),7.60–7.49(m,2h),7.44–7.28(m,5h),6.91–6.73(m,1h),5.97(d,j=8.5hz,1h),5.75(s,1h),4.65(t,j=9.7hz,1h),4.58–4.40(m,2h),4.00–3.89(m,2h),3.88–3.78(m,1h).13cnmr(101mhz,acetone-d6)δ157.07,140.19,138.95,135.32,130.80,129.70,128.87,127.37,125.66,124.06,116.93,102.39,98.86,82.53,69.21,69.06,67.93,58.02.;hrms(esi)calcdforc27h22ino7na[m+na]+622.0333,found622.0335.
将化合物7-2(500mg,0.83mmol)溶解于干燥得dmf(4ml)中。在冰浴和氮气保护下,加入nah(66.8mg,1.67mmol)。反应体系搅拌15min,加入bnbr(0.15ml,1.26mmol)。然后将体系升至室温,在此温度下搅拌3小时,点板监控反应完全,用二氯甲烷稀释,依次用水1nhcl、饱和nahco3、和饱和nacl洗,再用无水na2so4干燥,过滤旋去滤液上柱。用(pe/ea=10:1)过柱得到白色固体7-3(517.7mg,90%):[α]d25=+101(c0.5,chcl3);1hnmr(400mhz,acetone-d6)δ7.83(d,j=5.1hz,4h),7.66–7.58(m,3h),7.46–7.36(m,3h),7.36–7.26(m,2h),7.05–6.87(m,5h),6.77(td,j=7.4,1.7hz,1h),5.94(d,j=8.0hz,1h),5.83(s,1h),4.83(d,j=12.3hz,1h),4.64–4.44(m,4h),4.07–3.90(m,3h).13cnmr(101mhz,acetone-d6)δ139.20,138.02,134.25,129.79,128.74,128.04,127.90,127.74,127.30,126.18,124.74,123.16,115.97,101.05,97.84,82.37,74.78,73.64,68.10,66.48,55.19.hrms(esi)calcdforc34h28ino7na[m+na]+712.0803,found712.0778.
将化合物7-3(460mg,0.67mmol)溶解于干燥thf(7ml),加入bh3-nme3(200mg,2.7mmol),冷却至0℃,加入alcl3(360mg,2.7mmol),将体系移至室温,在此温度下搅拌3h,用ch2cl2稀释后,倒入1nh2so4中搅拌15分钟.有机相依次用水、饱和nahco3、饱和nacl洗、和用无水na2so4干燥.。过滤旋干滤液上柱,用(pe/ea=3:1)过柱得到白色固体7(381.8mg,81%):[α]d25=+44.5(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ7.81(brs,1h),7.65(brs,3h),7.57(dd,j=1.6,8.0hz,1h),7.34-7.26(m,5h),7.18-7.12(m,2h),7.09-7.06(m,2h),6.98-6.91(m,3h),6.70(ddd,j=2.4,6.8,8.0hz,1h),5.72(d,j=8.4hz,1h),4.79(d,j=12.0hz,1h),4.66-4.53(m,4h),4.36(dd,j=8.4,10.8hz,1h),3.93-3.80(m,4h);13cnmr(100mhz,cdcl3)δ156.1,139.2,138.1,137.8,133.9,129.6,128.6,128.3,128.0,127.9,127.8,127.6,124.6,123.4,116.2,97.8,97.7,86.8,78.8,74.7,74.6,73.8,73.5,70.3,55.1;hrms(esi)calcdforc34h30ino7na[m+na]+714.0959,found714.0964.
5、化合物10的合成参考文献hu,y.;yu,k.;shi,l.-l.;liu,l.;sui,j.-j.;liu,d.-y.;xiong,b.;sun,j.-s.j.am.chem.soc.2017,139,12736-12744.具体反应步骤如下:
将化合物10-1(190mg,0.34mmol)选择性上苄基得中间粗产物。将上述中间体化合物溶解于干燥的dmf(2ml)中。加入nah(27.4mg,0.68mmol,)在0℃和氮气保护下,体系在此温度下搅拌15min,加入allbr(35.2μl,0.4mmol)。然后体系被移至室温再搅拌2h。点板监控反应完全,用二氯甲烷稀释,依次用水、1nhc、饱和nahco3、和饱和nacl洗,再用无水na2so4干燥,过滤旋去滤液上柱。用(pe/ea=10:1)过柱得到白色固体10-2(193.4mg,76%for2steps):[α]d25=+82.2(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ7.75(dd,j=1.6,8.0hz,1h),7.52-7.22(m,11h),7.04(dd,j=1.2,8.0hz,1h),6.78(td,j=1.6,7.6hz,1h),6.02-5.92(m,1h),5.64(s,1h),5.52(d,j=2.0hz,1h),5.36(dq,j=1.6,17.2hz,1h),5.25(dq,j=1.2,10.0hz,1h),4.96(d,j=12.4hz,1h),4.81(d,j=12.0hz,1h),4.38-4.33(m,1h),4.30-4.27(m,2h),4.26-4.20(m,1h),4.18-4.14(m,1h),4.00(t,j=2.0hz,1h),3.90-3.82(m,2h);13cnmr(100mhz,cdcl3)δ154.7,139.5,138.5,137.7,137.6,134.7,129.6,129.0,128.5,128.3,128.1,127.8,126.2,124.3,118.2,114.9,101.6,98.0,87.4,78.9,76.6,75.4,73.5,73.4,68.6,65.4;hrms(esi)calcdforc29h30io6[m+h]+601.1082,found601.1078.
将化合物10-2(150mg,0.25mmol)溶解于ch2cl2(4ml)中,加入h2o(1ml)和tfa(2ml)在0℃下。体系在此温度下搅拌反应1h,用二氯甲烷稀释,再加三乙胺淬灭反应。依次用水、饱和nahco3、饱和nacl洗、和用无水na2so4干燥.。过滤旋干滤液的粗产物不需要进一步纯化。将上述粗产物经过简单的苄基化反应得到白色固体化合物10-3(142mg,82%for2steps):[α]d25=+36.5(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ7.74(d,j=1.6,8.0hz,1h),7.46-7.44(m,2h),7.36-7.17(m,15h),6.77(td,j=1.6,7.6hz,1h),6.00-5.91(m,1h),5.55(d,j=1.6hz,1h),5.35(dq,j=1.6,17.2hz,1h),5.23(dq,j=1.2,10.4hz,1h),4.93(d,j=10.8hz,1h),4.83(ab,2h),4.64(d,j=12.0hz,1h),4.53(d,j=10.8hz,1h),4.45(d,j=12.0hz,1h),4.28-4.18(m,3h),4.11(t,j=9.6hz,1h),3.97(dd,j=2.0,3.2hz,1h),3.86(ddd,j=2.0,4.4,9.6hz,1h),3.79(dd,j=4.4,10.8hz,1h),3.67(dd,j=1.6,10.8hz,1h);13cnmr(100mhz,cdcl3)δ155.3,139.3,138.5,138.4,138.3,134.9,129.7,128.6,128.4,128.3(2c),128.0,127.9,127.7,127.6,124.2,118.0,115.6,97.4(2c),87.5,79.4,75.2,74.9,74.6,73.3,72.9,72.7,72.4,69.0;hrms(esi)calcdforc36h37io6na[m+na]+715.1527,found715.1534.
将化合物10-3(120mg,0.17mmol)溶解于dcm/meoh(2ml,v/v=1:1)混合溶剂中,加入pdcl2(9.2mg,0.052mmol)在室温和氮气保护下。反应体系在此温度下搅拌6h,点板监控反应完全,用et3n淬灭反应,垫硅藻土和硅胶粉过滤,旋去滤液,过层析柱(pe/ea=5:1)得到无色糖浆化合物10(90mg,79%):[α]d25=+63.9(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ7.74(dd,j=1.6,7.6hz,1h),7.43-7.16(m,17h),6.77(td,j=1.6,7.6hz,1h),5.62(d,j=1.6hz,1h),4.87(d,j=10.8hz,1h),4.81(t,j=12.0hz,2h),4.61(d,j=12.0hz,1h),4.55(d,j=10.8hz,1h),4.44(d,j=12.0hz,1h),4.22-4.18(m,2h),4.04(dd,j=8.4,10.0hz,1h),3.87(ddd,j=2.0,4.4,10.0hz,1h),3.77(dd,j=4.4,10.8hz,1h),3.63(dd,j=2.0,10.8hz,1h),2.59(brs,1h);13cnmr(100mhz,cdcl3)δ155.0,139.4,138.3,138.1,137.8,129.7,128.7,128.4,128.3,128.2,128.0(2c),127.8,127.7,124.1,115.2,98.3,87.3,79.5,75.3,74.0,73.4,72.4,72.3,68.7,68.5;hrms(esi)calcdforc33h33io6na[m+na]+675.1214,found675.1221.
实施例1
(1)化合物3的合成
将化合物1(60mg,0.092mmol)和2(30.6mg,0.046mmol)溶于干燥的ch2cl2(1ml)中,加入活化后的4ams在n2的保护下。反应体系在室温下搅拌15分钟后,放入-40℃低温下继续搅拌5分钟,加入nis(49.4mg,0.221mmol)和tfoh(8.0μl,0.092mmol),在低温下再反应2小时,tlc点板监控反应完全,加入et3n淬灭反应.过滤、饱和碳酸氢钠、饱和nacl洗,无水硫酸钠干燥。过滤减压浓缩得粗产物,最后再柱层析(pe:ea:dcm=3:1:1)得到白色固体化合物3(49.7mg,90%).[α]d25=-3.0(c0.9,chcl3);1hnmr(400mhz,cdcl3)δ8.58(d,j=4.8hz,1h),8.05(dd,j=1.2,8.4hz,2h),7.74(td,j=1.2,7.6hz,1h),7.64(dd,j=1.6,7.6hz,1h),7.58-7.53(m,1h),7.44-7.13(m,16h),6.72(td,j=1.2,7.2hz,1h),6.05(dd,j=8.0,10.0hz,1h),5.51(dd,j=1.6,9.2hz,1h),5.46(d,j=14.0hz,1h),5.42-5.38(m,1h),5.23(d,j=13.6hz,1h),5.11-5.00(m,3h),4.75(d,j=11.6hz,1h),4.70(d,j=12.4hz,1h),4.57(d,j=12.4hz,1h),4.34(dd,j=2.4,12.8hz,1h),4.16(dd,j=4.8,12.4hz,1h),4.04-3.97(m,3h),3.83-3.77(m,2h),3.73(dd,j=5.6,10.0hz,1h),3.27(t,j=10.4hz,1h),2.88(dd,j=4.8,12.4hz,1h),2.11(s,3h),2.09(s,3h),2.05(s,3h),2.00(s,3h),1.84(t,j=12.4hz,1h);13cnmr(100mhz,cdcl3)δ170.8,169.9(2c),169.6,167.0,165.3,156.6,154.2,149.2,139.4,138.4,137.7,137.5,132.9,130.7,130.0,129.4,128.5,128.4,128.3,127.8,127.7,124.4,123.4,121.6,116.5,100.6,98.8,87.0,80.0,74.5,73.9,72.6,72.1,71.7,71.1,68.0,67.9,67.4,63.8,62.2,60.1,37.4,21.1,21.0(2c),20.8(2c);hrms(esi)calcdforc56h58in4o18[m+h]+1201.2785,found1201.2882.
(2)化合物3保护基的脱除
将化合物3(45.6mg,0.038mmol)溶于无水甲醇(2ml)中,加入cu(oac)2(7.5mg,0.042mmol).将反应体系升温至45℃,体系搅拌过夜,用et3n淬灭反应,减压浓缩,过层析柱得白色固体(pe/ea=2:1)得白色糖浆39(38.4mg,90%):[α]d25=+5.3(c1.0,chcl3);1hnmr(400mhz,cdcl3)δ8.04(dd,j=1.2,8.0hz,2h),7.65(dd,j=1.6,7.6hz,1h),7.58-7.54(m,1h),7.44-7.40(m,4h),7.36-7.32(m,2h),7.13(m,8h),6.74(td,j=1.6,7.6hz,1h),6.05(dd,j=8.0,10.0hz,1h),5.51(dd,j=1.6,9.2hz,1h),5.43(ddd,j=2.4,4.8,9.6hz,1h),5.08(d,j=8.0hz,1h),5.05(d,j=11.6hz,1h),4.87(ddd,j=4.8,9.6,12.0hz,1h),4.75(d,j=11.2hz,1h),4.70(d,j=12.0hz,1h),4.57(d,j=12.4hz,1h),4.35(dd,j=2.4,12.8hz,1h),4.16(dd,j=5.2,12.8hz,1h),4.04(d,j=2.8hz,1h),3.96(dd,j=6.8,10.4hz,1h),3.86-3.77(m,3h),3.74(s,3h),3.64(dd,j=6.8,10.0hz,1h),3.25(dd,j=9.6,10.4hz,1h),2.75(dd,j=4.4,12.4hz,1h),2.17(s,3h),2.12(s,3h),2.09(s,3h),2.01(s,3h),1.79(t,j=12.8hz,1h);13cnmr(100mhz,cdcl3)δ170.9,169.9(2c),169.7,167.6,165.3,156.6,139.4,138.4,137.6,133.0,130.7,130.1,129.4,128.5,128.4,128.2,127.8,127.7,124.4,116.6,100.6,98.8,87.1,80.0,74.5,73.7,72.4,72.1,71.8,71.1,71.0,68.0,67.8,63.5,62.3,60.0,53.2,37.4,21.2,21.0,20.9,20.8;hrms(esi)calcdforc51h54in3o18na[m+na]+1146.2339,found1146.2359.
(3)化合物4保护基的转化
将化合物4(39.3mg,0.035mmol)溶于干燥的吡啶(0.6ml)中,加入硫代乙酸(0.6ml)在0℃和n2保护下.反应体系移至室温搅拌24h.减压旋干,过层析柱(pe/ea=1:3)得无色液体化合物5(28.7mg,72%):[α]d25=+7.0(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ8.05(dd,j=1.2,8.4hz,2h),7.64(dd,j=1.6,8.0hz,1h),7.57-7.53(m,1h),7.45-7.40(m,4h),7.36-7.32(m,3h),7.29-7.13(m,7h),6.73(td,j=1.6,7.6hz,1h),6.04(dd,j=8.0,10.0hz,1h),5.45(ddd,j=2.8,6.4,8.8hz,1h),5.32-5.29(m,2h),5.12(d,j=8.0hz,1h),5.05(d,j=11.2hz,1h),4.92(ddd,j=4.4,9.6,12.0hz,1h),4.78-4.68(m,2h),4.57(d,j=12.4hz,1h),4.38(dd,j=2.4,12.4hz,1h),4.17-4.06(m,3h),4.05(dd,j=6.4,12.4hz,1h),3.92-3.87(m,2h),3.84(dd,j=2.8,10.4hz,1h),3.72(s,3h),3.71(dd,j=1.6,11.6hz,1h),2.64(dd,j=4.8,13.2hz,1h),2.17(s,3h),2.10(s,3h),2.05(s,3h),2.00(t,j=12.4hz,1h),1.94(s,3h),1.89(s,3h);13cnmr(100mhz,cdcl3)δ171.1,170.9,170.5,170.4,170.1,168.2,165.4,156.6,139.3,138.6,137.7,132.9,130.7,130.1,129.5,128.4,128.3,128.2,127.8,127.7,127.6,124.4,116.6,100.6,99.3,87.0,80.1,74.6,73.5,72.8,72.3,72.1,71.2,69.0,68.5,67.5,63.2,62.9,53.1(2c),49.4,38.0,23.3,21.2,21.0,20.9;hrms(esi)calcdforc53h58ino19na[m+na]+1162.2540,found1162.2574.
(4)化合物5的ip保护基进行活化
将化合物5(38mg,0.033mmol),ph3p(3.8mg,0.014mmol),pd(pph3)2cl2(2.2mg,0.003mmol),和cui(2.8mg,0.015mmol)溶解在dmf(0.26ml)和ipr2nh(0.87ml)的混合溶剂中,在氮气保护下,降温到-78℃后用真空隔膜泵换气。重复三次换气后加入溶解对甲氧基苯乙炔(4μl,0.03mmol)的dmf(0.26ml)。升温至70℃后反应4h。点板监控反应完全加入nh4cl淬灭反应,用乙酸乙酯稀释后垫硅藻土和硅胶粉过滤,先用饱和的nh4cl洗三次后,再用饱和的nacl洗两次合并有机相。用无水的na2so4干燥。垫上硅胶粉过滤,滤液旋干后装柱。用(pe/ea=1:3)过柱得到淡黄色固体6(33.2mg,87%):[α]d25=-37.5(c0.64,chcl3);1hnmr(400mhz,cdcl3)δ7.83(dd,j=1.2,8.0hz,2h),7.69(dd,j=7.2,12.0hz,1h),7.57(t,j=7.2hz,1h),7.49-7.38(m,6h),7.34-7.30(m,4h),7.27-7.12(m,6h),6.94(t,j=7.6hz,1h),6.83(d,j=8.8hz,2h),6.09(dd,j=8.0,10.0hz,1h),5.47-5.38(m,2h),5.31(d,j=8.4hz,1h),5.25(d,j=8.0hz,1h),5.05(d,j=11.2hz,1h),4.93(td,j=4.4,11.2hz,1h),4.74(d,j=11.2hz,1h),4.70(d,j=12.0hz,1h),4.57(d,j=12.0hz,1h),4.40(dd,j=1.6,12.4hz,1h),4.20-4.07(m,3h),4.05(dd,j=6.8,12.4hz,1h),3.98(t,j=7.2hz,1h),3.89-3.85(m,2h),3.83(s,3h),3.76-3.69(m,1h),3.72(s,3h),2.64(dd,j=4.4,13.2hz,1h),2.17(s,3h),2.11(s,3h),2.05(s,3h),2.01(t,j=12.4hz,1h),1.93(s,3h),1.89(s,3h);13cnmr(100mhz,cdcl3)δ171.0,170.8,170.5,170.3,170.0,168.2,165.2,159.4,157.7,138.7,137.8,133.4,132.9,132.6,132.2,132.1,130.2,129.9,129.1,128.7,128.4,128.3,128.1,128.0,127.7,127.6,127.5,122.0,115.9,115.2,114.2,113.6,99.8,99.4,93.9,83.6,80.4,74.5,73.4,72.8,72.4,72.1,71.4,69.0,68.5,67.6,63.4,63.0,55.4,53.1,49.4,38.0,23.3,21.2,21.0,20.9,20.8;hrms(esi)calcdforc62h65no20na[m+na]+1166.3992,found1166.4021.
(5)化合物8的合成
将化合物6(59.6mg,0.052mmol)和7(72mg,0.104mmol)溶解于活化aw-300ms干燥的二氯甲烷(1ml)中,在室温和氮气保护下搅拌30min.。降温至-35℃,加入nis(15.6mg,0.078mmol)和tmsotf(4.7μl,0.026mmol)。反应3.5h后,加入et3n淬灭反应。过滤旋干滤液上柱,用(pe/ea=1:2)过柱得到白色泡沫状糖苷化产物8(69.8mg,83%):[α]d25=+26.3(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ8.04(dd,j=1.2,8.4hz,2h),7.76-7.57(m,5h),7.52-7.44(m,3h),7.42-7.13(m,15h),7.05-7.00(m,4h),6.86-6.82(m,3h),6.65-6.61(m,1h),5.70(dd,j=7.6,10.0hz,1h),5.57(d,j=8.4hz,1h),5.34-5.32(m,3h),5.02(d,j=11.2hz,1h),4.94(d,j=12.4hz,1h),4.88-4.84(m,1h),4.83(d,j=8.0hz,1h),4.69(dd,j=4.8,12.4hz,1h),4.55-4.44(m,4h),4.38(dd,j=8.0,10.8hz,1h),4.29-4.25(m,2h),4.13-4.03(m,4h),4.01(dt,j=2.8,12.4hz,1h),3.82(dd,j=4.8,8.8hz,1h),3.73-3.54(m,6h),3.61(s,3h),2.54(dd,j=4.4,12.8hz,1h),2.13(s,3h),2.06(s,3h),2.04(s,3h),2.03(s,3h),1.92(t,j=12.4hz,1h),1.88(s,3h);13cnmr(100mhz,cdcl3)δ171.0,170.8,170.5,170.2,170.0,167.9,165.2,156.1,139.1,138.9,138.8,138.4,137.8,133.8,133.3,130.1,130.0,129.6,128.6,128.4,128.2(2c),128.0,127.8,127.7(2c),127.6,127.5,127.3,127.0,124.5,123.3,116.2,101.1,99.2,97.6,86.8,80.0,78.1,75.0,74.9,74.3,73.2,73.0,72.8,72.3,71.6,69.2,68.9,68.4,67.6,62.9,62.5,55.4,53.0,49.5,37.6,23.3,21.2,21.0,20.9(2c);hrms(esi)calcdforc81h83in2o25na[m+na]+1633.4222,found1633.4260.
(6)化合物8的ip保护基进行活化
实验操作于合成化合物6相同,得淡黄色固体化合物9(25mg,83%):[α]d25=-28.7(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ8.04(dd,j=1.2,8.4hz,2h),7.62-7.58(m,1h),7.48(dd,j=7.2,8.0hz,3h),7.37(dd,j=1.6,8.0hz,3h),7.30-7.13(m,18h),7.03-6.98(m,4h),6.87-6.79(m,6h),5.70(dd,j=7.6,10.0hz,1h),5.65(d,j=8.4hz,1h),5.32-5.25(m,3h),5.02(d,j=11.6hz,1h),4.93(d,j=12.4hz,1h),4.87-4.81(m,2h),4.69(d,j=12.4hz,1h),4.67(d,j=11.6hz,1h),4.53-4.48(m,4h),4.39(dd,j=8.4,10.8hz,1h),4.29-4.24(m,2h),4.12-4.02(m,4h),4.00-3.95(m,1h),3.85(s,3h),3.80(dd,j=4.8,8.4hz,1h),3.72-3.53(m,6h),3.60(s,3h),2.53(dd,j=4.4,12.8hz,1h),2.13(s,3h),2.06(s,3h),2.033(s,3h),2.030(s,3h),1.92(t,j=13.6hz,1h),1.88(s,3h);13cnmr(100mhz,cdcl3)δ171.1,170.8,170.4,170.2,170.0,167.9,165.2,159.4,157.3,139.0,138.9,138.5,137.8,133.3(2c),133.0,131.5,130.1,130.0,129.2,128.6,128.4(2c),128.2,128.1,127.9,127.8,127.7,127.6(2c),127.5,127.3,126.9,123.1,122.3,115.7,115.2,114.0,113.7,101.1,99.2,97.0,93.3,83.5,80.0,78.2,77.7,75.1,74.8,74.3,73.2,73.0,72.8,72.3,71.6,69.2,68.8,68.3,67.6,62.8,62.4,55.4,53.0,49.5,37.5,23.3,21.2,21.0(2c),20.9;hrms(esi)calcdforc90h90n2o26k[m+k]+1653.5413,found1653.5417.
(7)化合物11的合成
实验操作参照化合物8的合成,得白色泡沫状糖苷化产物11(32mg,82%)[α]d25=+27.5(c0.5,chcl3);1hnmr(400mhz,cdcl3)δ8.00(d,j=7.6hz,2h),7.66-7.56(m,4h),7.50-7.43(m,4h),7.36-7.31(m,5h),7.24-7.06(m,24h),7.03-6.95(m,4h),6.85-6.80(m,3h),6.70-6.65(m,2h),5.68(dd,j=8.0,10.4hz,1h),5.35-5.30(m,2h),5.22(dd,j=2.8,5.6hz,1h),5.13(d,j=2.0hz,1h),5.01(d,j=11.6hz,1h),4.89-4.75(m,8h),4.68(d,j=3.2hz,1h),4.65(d,j=2.4hz,1h),4.55-4.67(m,4h),4.37(d,j=10.8hz,1h),4.28-4.19(m,4h),4.12-3.97(m,6h),3.87-3.83(m,3h),3.69-3.50(m,7h),3.56(s,3h),3.31(d,j=10.8hz,1h),2.92(dd,j=5.6,11.2hz,1h),2.54(dd,j=4.8,12.8hz,1h),2.13(s,3h),2.06(s,3h),2.03(s,3h),2.02(s,3h),1.92(t,j=12.0hz,1h),1.88(s,3h);13cnmr(100mhz,cdcl3)δ171.1,170.9,170.5,170.2,170.1,167.9,165.2,154.9,139.2,138.9,138.8,138.5,138.4,138.1,137.8,133.6,133.3,131.8,130.0(2c),129.4,128.6,128.5(2c),128.4(2c),128.3(2c),128.2(2c),128.1,128.0,127.8,127.7,127.6(2c),127.4(2c),127.3,127.0,124.0,123.2,123.0,114.8,101.0,99.1,97.0,95.8,87.2,80.0,78.2,77.7,75.0,74.9,74.8,74.4,74.2,73.4(2c),73.0,72.8,72.7,72.6(2c),72.4,71.6,70.7,69.7,69.1,69.0,68.8,67.6,62.9,62.4,55.7,53.0,49.4,37.5,23.3,21.2,21.0(2c),20.9;hrms(esi)calcdforc108h112in2o30[m+h]+2044.6373,found2044.6370.
实施例2
(1)反应参考实施例1中步骤(1)的步骤和参数,所不同的是糖基给体化合物1、糖基受体化合物2、nis和tfoh的摩尔比为2:1:5:5,所述化合物1在第一溶剂中的浓度为0.003mol/l;所述糖苷化反应温度为-20℃;全α构型产率87%
(2)反应参考实施例1中步骤(2)的步骤和参数,所不同的是化合物3与醋酸铜的摩尔比1:4,所述化合物3在第二溶剂中的浓度优0.004mol/l,溶剂为甲醇和二氯甲烷,温度50℃,产率60%
(3)反应参考实施例1中步骤(3)的步骤和参数,所不同的是所述化合物4与硫代乙酸的摩尔比优选为1:10,以体积比为1:1的二氯甲烷和吡啶为溶剂。所述化合物4在溶剂中的浓度为0.001,产率60%。
(4)反应参考实施例1中步骤(4)的步骤和参数,所不同的是将所述化合物5、ph3p、pd(pph3)2cl2、cui和对甲氧基苯乙炔的摩尔比为1:0.5:0.2:0.5:1.6,化合物5在第四溶剂中的浓度为0.01,产率为70%。
(5)反应参考实施例1中步骤(5)的步骤和参数,所不同的是所述化合物7和路易斯酸、nis的摩尔比为1:0.5:1.5;化合物6和化合物7的摩尔比为1:1;糖苷化给体与第六有机溶剂的质量体积比为20mg/ml;产率70%。
(6)反应参考实施例1中步骤(6)的步骤和参数,所不同的是所述化合物8、ph3p、pd(pph3)2cl2、cui和对甲氧基苯乙炔的摩尔比为1:0.45:0.1:0.45:1.5;化合物8在第七有机溶剂中的浓度为0.1mol/l,产率60%。
(7)反应参考实施例1中步骤(7)的步骤和参数,所不同的是所述化合物9和路易斯酸、nis的摩尔比为1:0.5:1.5;化合物9和化合物10的摩尔比为1:1;式i所示的糖苷化给体与第六有机溶剂的质量体积比20mg/ml;产率65%。
对比例1:步骤(1)中,将化合物1(60mg,0.092mmol)和2(30.6mg,0.046mmol)溶于干燥的ch2cl2(1ml)中,加入活化后的4ams在n2的保护下。反应体系在室温下搅拌15分钟后,放入-40℃低温下继续搅拌5分钟,加入nis(49.4mg,0.221mmol)和tfoh(1.6μl,0.0184mmol),在低温下再反应2小时,tlc点板监控反应完全,加入et3n淬灭反应.过滤、饱和碳酸氢钠、饱和nacl洗,无水硫酸钠干燥。过滤减压浓缩得粗产物,最后再柱层析(pe:ea:dcm=3:1:1)得到白色固体化合物3(5mg,9%)
对比例2:步骤(2)中,将化合物3(45.6mg,0.038mmol)溶于无水甲醇(0.5ml)和二氯甲烷(1.5ml)中,加入cu(oac)2(7.5mg,0.042mmol).将反应体系升温至45℃,体系搅拌过夜,用et3n淬灭反应,减压浓缩,过层析柱得白色固体(pe/ea=2:1)得白色糖浆4(25.6mg,60%):
对比例3:步骤(3)中,将化合物4(39.3mg,0.035mmol)溶于干燥的吡啶(0.3ml)和二氯甲烷(1ml)中,加入硫代乙酸(0.6ml)在0℃和n2保护下.反应体系移至室温搅拌24h.减压旋干,过层析柱(pe/ea=1:3)得无色液体化合物5(23.9mg,60%)。
- 还没有人留言评论。精彩留言会获得点赞!