土壤三域微生物高通量绝对定量方法与流程
本发明属于微生物领域,涉及一种以高通量测序技术为基础,通过样品中三域通用内标菌株的添加和利用,实现三域微生物(古菌、细菌、真菌)高通量绝对定量的方法。
背景技术:
微生物是个体难以用肉眼观察的一切微小生物的统称,因为具有适应性强、易产生变异、可适应环境多样、种类丰富等特点,彰显出了巨大的开发潜能,广受医药、化工、农业、生态环境等各界学者关注。研究微生物多样性,探究微生物群落分布模式及变化机制,对于加深理解环境变化下微生物群落演替及响应机制并对其加以利用具有重要意义。
地球环境可分为五大圈层:大气圈、岩石圈、生物圈、水圈、土壤圈。作为五大圈层中与其他圈层交互最为频繁的界面,土壤圈具有最为复杂的内部结构,是地球上异质性极为明显的自然体,为微生物提供了复杂多样的生存环境,也因此涵盖了地球上数量最为庞大、种类最为丰富的微生物群体。
基于16s或18srrna/its基因序列的相似性差异,微生物可被划分为古菌域、细菌域和真菌域。环境中的三域微生物各有其优势:古菌可在于极端环境中生存,细菌种类丰富,真菌生物量大。揭示复杂的环境中三域微生物如何各司其职维持生态的平衡,古菌、细菌、真菌物种间如何竞争或互利共生,对于评价及进一步利用其生态功能具有重要意义。
纵观微生物群落结构研究方法发展史,研究手段经历了从传统的平板分离培养到深入探究微生物dna分子水平的革新,微生物群落分子水平的研究方法克服了传统分离培养方法的局限,使得对微生物群落结构的解析更加深入和透彻。特别是高通量测序技术的出现,极大程度地推动了学者对微生物群落的理解:通过对微生物细胞内特定遗传物质的测定(16srrna或18srrna/its基因片段),样本中微生物群落结构的多样性信息和含量组成可被进一步解析。
然而,就目前基于高通量测序技术的研究中,古菌域、细菌域、真菌域微生物是分域进行测序分析的,单个域内的优势物种及其相对丰度组成可被解析,但古菌、细菌、真菌微生物各自绝对含量的信息尚且缺乏,使得微生物群落结构组成信息“定性却又无法精确定量”,直接导致了三域微生物间无法相互比较,并进一步限制了基于微生物数量生态理论对三域微生物的探索。现有高通测序技术分析结果中,均以相对丰度的形式表征微生物的组成,在分析描述微生物变化时存在一定的问题:
一、当聚焦单域微生物变化时,相对丰度的描述形式可用于表征单个样品中不同分类单元的优势程度差异,而比较同一分类单元在不同样本中变化情况,特别是样本间微生物总量差异较大时,相对丰度的结果可能会导致错误的结论。该问题已由专利《土壤细菌高通量绝对定量方法》(专利号:201710296291.7)进行校正解决。
二、单个样品中,针对于各域的高通量测序结果只能孤立地表征其中一个域中不同分类单元的优势程度,而从属于古菌域、真菌域、细菌域的不同分类单元(taxon)三域间的垮域比较难以实现。
三、当我们试图比较不同样本中不同域的分类单元的数量差异时,因域间及样本间本身存在的绝对总量的差异,若误用相对丰度来进行比较表征,将可能导致错误的结论。
为克服传统高通量测序结果中相对丰度表征的缺陷,以微生物数量生态学为基础、全面统筹地研究三域微生物间的相互关系及其对环境变化的响应,急需开发一种实现古菌、细菌、真菌三域微生物高通量绝对定量的技术,以恒定的指标(绝对含量)实现三域微生物的跨样本跨域相互比较,弥补现有方法的缺陷,对明确环境中古菌、细菌、真菌各自真实的数量,阐明微生物响应机理全方位理解生物变化,及进一步研究微生物生态学具有十分重要的意义。
技术实现要素:
本发明要解决的技术问题是提供一种土壤三域微生物(古菌、细菌、真菌)高通量绝对定量方法;本发明同时解决高通量测序结果中采用相对丰度用于单样本跨域及跨样本垮域比较中的存在的缺陷,提供技术手段实现三域微生物绝对含量表征,从而达到三域微生物间可以跨样本跨域相互比较的目的。该技术利用一株基因改造的三域通用内标菌株,实现三域微生物的高通量绝对定量,为推进三域微生物为整体的群落研究提供技术支持。
为了解决上述技术问题,本发明提供一种土壤三域微生物(古菌、细菌、真菌)高通量绝对定量方法,选用保藏编号为cctccno:m2017770的大肠杆菌escherichiacoliwyl(e.coliwyl)作为三域通用内标菌株,包括以下步骤:
1)、将三域通用内标菌株e.coliwyl,重悬于无菌水中,得od600nm为0.9~1.0的菌悬液;
2)、土壤样品中三域通用内标菌株的添加:
吸取200μl步骤1)所得的菌悬液至2g土壤中搅拌混匀(从而实现三域通用内标菌株在土壤中终浓度为5×106copiesg-1左右);
注:该过程于冰浴环境下完成,以防止搅拌过程中微生物生长导致的群落结构变化;使用无菌玻璃棒搅拌混匀,搅拌时间约为5min,
3)、土壤样品dna提取;
4)、样品中三域通用内标菌株绝对定量:
采用spec-f/r引物(引物碱基信息如表2所示)对土壤样品中三域通用内标菌株进行定量,反应体系(20μl)为:10μlsybrgreenqpcrsupermix-udgwithrox(invitrogen,carlsbad,usa),正、反向引物各0.4μl,8.2μl无核酸酶水,1μl染色体dna;反应条件如下所示:
表1实时荧光定量pcr反应条件
5)土壤样品高通量测序;
6)、三域微生物绝对含量计算。
作为本发明的土壤三域微生物(古菌、细菌、真菌)高通量绝对定量方法的改进:
所述步骤5)为:
采用通用引物515f/806r测定古菌和细菌微生物群落结构组成,并获得三域通用内标菌株在古菌和细菌微生物群落中的相对丰度;采用通用引物its-f/r测定真菌微生物群落结构组成,并获得三域通用内标菌株在真菌微生物群落中的相对丰度(引物碱基信息如表2所示);
所获得高通量测序数据采用qiime软件(version1.9.0)进行质控与过滤;正向与反向测序结果采用flash软件进行拼接;采用uparse软件将优质序列按97%的相似度归为一个otu(operationaltaxonomicunit);
将三域通用内标菌株中人工设计515f/806r及its-f/r所对应碱基信息分别添加进silva数据库及unite数据库,采用包含三域通用内标菌株碱基信息的silva数据库及unite数据库分别对原核微生物及真核微生物otu进行注释。
所述步骤6)为:
结合三域通用内标菌株的绝对含量aauiss及其在古菌和细菌微生物群落中相对丰度raa+b_uiss获得古菌与细菌微生物绝对总量taa+b(taa+b=aauiss/raa+b_uiss);古菌与细菌微生物群落结构中,其他各水平(门、纲、目、科、属)分类单元绝对含量可通过绝对含量与该分类单元在原核微生物群落中相对风度的乘积计算获得(aaa+b_taxon
=taa+b×rataxon);
同理,结合三域通用内标菌株的绝对含量aauiss及其在真菌微生物群落中相对丰度raf_uiss获得真菌微生物绝对总量taf(taf=aauiss/raf_uiss),真核微生物群落结构中,其他各水平(门、纲、目、科、属)分类单元绝对含量可通过绝对含量与该分类单元在真核微生物群落中相对风度的乘积计算获得(aaf_taxon=taf×rataxon)。
注:根据silva数据库及unite数据库的注释信息,将测序结果划分为古菌、细菌、真菌,至此,我们便获得了三域微生物的绝对含量,该数值为恒定指标,不会因不同样本生物量的差异而使得跨域、跨样本的比较出现偏差。
作为本发明的土壤三域微生物(古菌、细菌、真菌)高通量绝对定量方法的进一步改进:
步骤3):参照mobio
作为本发明的土壤三域微生物(古菌、细菌、真菌)高通量绝对定量方法的进一步改进,三域通用内标菌株的培养为:
吸取200μl甘油(25%)-80℃保存的三域通用内标菌株于20mllb培养基中,于37℃250rpm条件下培养14-16h后,4℃6000×g离心获得菌体,弃上清,并采用无菌水将菌体重悬浮,再次4℃6000×g离心,弃上清后重复无菌水重悬再离心步骤,最后将三域通用内标菌株重悬于无菌水中,得od600nm为0.90~1.0的菌悬液。
在本发明的步骤2)中,采用200ul菌液加入2g土壤,可以节省土壤样品量,当整体土壤采样量小时(例如土钻采样时),减小三域微生物高通量绝对定量测试分析样本量,有利于在测定土壤各项理化性质的同时实现绝对定量,获得更为全面的土壤理化及微生物信息。
本发明所指三域通用内标菌株为escherichiacoliwyl(e.coliwyl),保藏于中国典型培养物保藏中心(chinacenterfortypeculturecollection,cctcc),保藏编号为m2017770。实验室条件采用25%甘油于-80℃条件保存。该保藏编号为cctccno:m2017770的大肠杆菌escherichiacoliwyl(e.coliwyl)在申请号2019100653049的《校正高通量测序原核与真核微生物基因序列读取数的方法及所用菌》中有明确告知。
表2高通量测序及qpcr引物信息
相对于现有技术而言,本发明具有如下技术优势:
突破因等同测序深度导致的域间及样本间数量差异被模糊、微生物间只能以相对丰度表征而无法进行跨样本跨域比较的限制,实现了三域微生物的高通量定量。实验操作简单,在原有高通量测序手段的基础上,仅需添加一株三域通用内标菌株,即可获得准确可靠的三域微生物(古菌、细菌、真菌)高通量绝对定量测定结果。分析结果中,三域微生物数量以绝对含量(copiesg-1)形式表征,准确恒定,更有利于后续分析,并提供了一种更为全方位的视角统筹研究三域微生物间的相互作用关系。
专利《校正高通量测序原核与真核微生物基因序列读取数的方法及所用菌》中所提供方法也可实现单样本跨域、跨样本跨域的比较,但仅限于在30,000-50,000条的测序深度下,用序列读取数(numberofreads)表征古菌、细菌、真菌的多少从而进行比较,而其实际在土壤中的绝对含量不得而知。本发明中的方法不仅可以实现单样本跨域、跨样本跨域的比较,还可获得古菌、细菌、真菌在土壤中真实的绝对含量,结果以copiesg-1为单位计。
附图说明
下面结合附图对本发明的具体实施方式作进一步的详细说明。
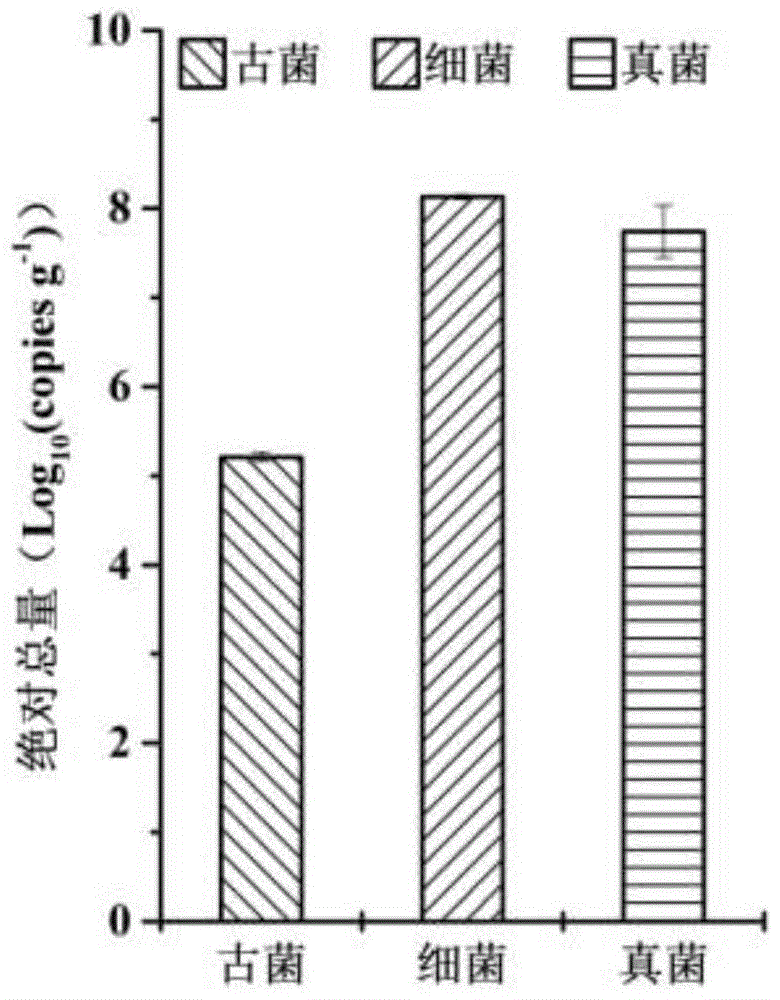
图1为土壤样品a中古菌、细菌和真菌的绝对总量;
图2为土壤样品a中奇古菌门、迷踪菌门和球囊菌门的相对丰度及绝对含量;
图3为土壤样品a和b门水平相对丰度及绝对含量;
图4为土壤样品a中子囊菌门和土壤样品b中变形菌门的相对丰度及绝对含量比较结果。
具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
实施例1、通过三域通用内标菌株实现同一土壤样品中跨域比较,依次进行以下步骤:
(1)将保存于-80℃的三域通用内标菌株e.coliwyl取出,吸取200μl于20ml灭菌液体lb培养基中,于37℃250rpm条件下培养14-16h。
(2)将步骤(1)所得的菌液转移至无菌的50ml离心管中,6000×g条件下离心5min获得菌体,弃上清。
(3)先添加5ml4℃预冷的无菌水至离心管,将菌体重悬,再添加25ml无菌水并混合均匀。
(4)将离心管置于高速离心机中,6000×g条件下离心5min获得菌体,弃上清。
(5)重复(3)-(4)步骤两次。
(6)将所获得菌体重悬于无菌水中,并调整菌悬液吸光度od600nm至0.9左右。
(7)吸取200μl步骤(6)所得的菌悬液于2g土壤中,将盛装土壤样品的容器置于冰浴中,使用无菌玻璃棒搅拌5min,使得三域通用内标菌株与土壤样品混合均匀。
(8)称取0.25g含有三域通用内标菌株的土壤,采用mobio
(9)采用spec-f/r引物对土壤dna中e.coliwyl进行qpcr绝对定量,获得三域通用内标菌株的绝对含量aauiss。
采用spec-f/r引物对土壤样品中三域通用内标菌株进行定量,反应体系(20μl)为:10μlsybrgreenqpcrsupermix-udgwithrox(invitrogen,carlsbad,usa),正、反向引物各0.4μl,8.2μl无核酸酶水,1μl染色体dna(步骤(8)所得)。反应条件及相关引物信息如表1和表2所示。
熔解性曲线分析是qpcr绝对定量中确定实时荧光定量pcr反应产物特异性的手段:通过同时对目标dna与标准品质粒dna的特征性熔解温度进行测定,来确定产物的基因型是否与标准品质粒一致。此为公知的常规技术,相关参数设定可参照罗氏公司《
(10)分别采用引物515f/806r和its-f/r对同一土壤dna样品分别进行原核微生物(古菌和细菌)群落高通量测序和真核(真菌)微生物高通量测序,测序方法分别参照batesetal(2011)和tedersooetal(2014)文献报道方法进行,高通量测序引物碱基信息如表2所示。
(11)将步骤(10)所得的测序数据进行过滤分析(采用qiime软件(version1.9.0))。剔除测序长度<200bp、模糊碱基>6bp、聚合体>6bp、引物与接头中错配碱基≥1bp、质量低于q20、具有嵌合体的测序片段;正向与反向测序结果采用flash软件进行拼接(
(12)三域通用内标菌株e.coliwyl,其染色体基因片段上包含如下基因片段(方框标记碱基为its-f/its-r引物结合位点,下划线标记碱基为515f/806r引物结合位点):
将三域通用内标菌株中人工设计515f/806r及its-f/r所对应碱基信息分别添加进silva数据库(https://www.arb-silva.de/)及unite数据库(https://unite.ut.ee/),标记为e.coliwyl,采用包含校正菌株碱基信息的silva数据库及unite数据库分别对古菌和细菌otu及真菌otu进行注释,该注释参照
(13)首先采用公式计算土壤中古菌和细菌的绝对总量(taa+b)、真菌的绝对总量(taf):
taa+b=aauiss/raa+b_uiss
taf=aauiss/raf_uiss
获得绝对总量数据后,再根据古菌和细菌微生物群落各分类水平下(界、门、纲、目、科、属)各分类单元相对丰度组成(raa+b_taxon)及真菌微生物群落its基因各分类水平下(界、门、纲、目、科、属)各分类单元相对丰度(raf_taxon)组成计算其绝对含量:
aaa+b_taxon=taa+b×raa+b_taxon
aaf_taxon=taf×raf_taxon
至此,我们便获得了古菌、细菌、真菌各分类单元的绝对含量,可用于单样本跨域的比较。
通过上述实施例1的方法,测得三域通用内标菌株在古菌和细菌微生物群落中相对丰度raa+b_uiss为(4.63±1.44)%,在真菌微生物群落中相对丰度raf_uiss为(10.86±4.58)%,结合实时荧光定量pcr结果的三域通用内标菌株绝对含量aauiss为(6.56×106±1.77×106copiesg-1),可计算出土壤土著古菌和细菌的微生物总量(taa+b-aauiss)=aauiss/raa+b_uiss-aauiss=(6.56×106±1.77×106copiesg-1)/(4.63±1.44%)-(6.56×106±1.77×106copiesg-1)copiesg-1=1.36×108±7.70×106copiesg-1,根据注释信息,我们可获得土著古菌和细菌绝对含量分别为1.63×105±1.89×104copiesg-1和1.36×108±7.68×106copiesg-1;土壤土著真菌微生物总量(taf-aauiss)=aauiss/raf_uiss-aauiss=(6.56×106±1.77×106copiesg-1)/(10.86±4.58%)-(6.56×106±1.77×106copiesg-1)=6.32×107±3.39×107copiesg-1,如图1所示。
根据结果可以发现,古菌(105)、细菌(108)、真菌(107)的绝对含量(单位:copiesg-1)是存在数量级差异的,若以相对丰度作为比较标准,则有可能导致错误的结论。例如,奇古菌门和迷踪菌门在古菌和细菌微生物群落中的相对丰度为0.11±0.01%和0.12±0.02%,球囊菌门在真菌微生物群落中相对丰度为0.29±0.09%,若以相对丰度为比较,球囊菌门的相对丰度含量是显著高于奇古菌门和迷踪菌门的绝对含量的。但计算三者绝对含量发现,奇古菌门绝对含量=(6.56×106±1.77×106copiesg-1)/(4.63±1.44%)×(0.11±0.01%)=1.63×105±1.89×104copiesg-1,迷踪菌门绝对含量=(6.56×106±1.77×106copiesg-1)/(4.63±1.44%)×(0.12±0.02%)=1.77×105±2.11×104copiesg-1,球囊菌门绝对含量=(6.56×106±1.77×106copiesg-1)/(10.86±4.58%)×(0.29±0.09%)=2.09×105±1.24×105copiesg-1。通过对比发现,三个菌门的绝对含量实质上无显著差异,而只是其优势程度显著不同。绝对含量的缺失模糊了其种群大小的信息,使其无法跨域比较,而通过本发明所介绍的三域高通量绝对定量的方法能很好的弥补高通量测序手段的缺陷。
对比例1、无三域通用内标菌株添加单个土壤样品中三域微生物跨域的比较
不添加三域通用内标菌株,直接对单个土壤样品进行dna提取,并分别对其古菌域和细菌域、真菌域进行高通量测序,相关实验条件如测序条件、分析方法等均与实施例1保持一致。
所得结果为:因缺失三域通用内标菌株的添加,无法结合其相对丰度和绝对含量计算土壤样品中古菌和细菌及真菌的绝对总量,古菌、细菌、真菌各自微生物数量差异不得而知,进而无法对各域微生物中各分类单元的数量进行跨域比较,微生物间的相互比较仅限于域内及其优势程度,进一步限制了土壤三域微生物的统筹分析。
实施例2、通过三域通用内标菌株实现两个土壤样品中跨样本跨域比较
(1)将保存于-80℃的三域通用内标菌株e.coliwyl取出,吸取200μl于20ml灭菌液体lb培养基中,于37℃250rpm条件下培养14-16h。
(2)将(1)中菌悬液转移至无菌的50ml离心管中,6000×g条件下离心5min获得菌体,弃上清。
(3)先添加5ml4℃预冷的无菌水至离心管,将菌体重悬,再添加25ml无菌水并混合均匀。
(4)将离心管置于高速离心机中,6000×g条件下离心5min获得菌体,弃上清。
(5)重复(3)-(4)步骤两次。
(6)将所获得菌体重悬于无菌水中,并调整菌悬液吸光度od600nm至0.970左右。
(7)分别吸取200μl(6)中菌悬液于2g土样a和2g土样b中,将盛装土壤样品的容器置于冰浴中,使用无菌玻璃棒搅拌5min,使得三域通用内标菌株与土壤样品混合均匀。
(8)称取0.25g含有三域通用内标菌株的土壤,采用mobio
以下步骤(9)~步骤(13)针对土样a和土样b分别进行:
(9)采用spec-f/r引物对土样a和b的土壤dna中e.coliwyl进行qpcr绝对定量,获得三域通用内标菌株的绝对含量aauiss。
(10)采用引物515f/806r和its-f/r同时对土样a和b的土壤dna样品分别进行原核微生物(古菌和细菌)群落高通量测序和真核(真菌)微生物高通量测序,测序方法分别参照batesetal(2011)和tedersooetal(2014)文献报道方法进行。
(11)将测序数据进行过滤分析。剔除测序长度<200bp、模糊碱基>6bp、聚合体>6bp、引物与接头中错配碱基≥1bp、质量低于q20、具有嵌合体的测序片段;正向与反向测序结果采用flash软件进行拼接(
(12)将三域通用内标菌株中人工设计515f/806r及its-f/r所对应碱基信息分别添加进silva数据库(https://www.arb-silva.de/)及unite数据库(https://unite.ut.ee/),标记为e.coliwyl,采用包含校正菌株碱基信息的silva数据库及unite数据库分别对古菌和细菌otu及真菌otu进行注释,该注释参照(
(13)土壤样本中古菌和细菌的绝对总量(taa+b)、真菌的绝对总量(taf)计算方法与实施例1相同,结合三域内标菌株在原核(古菌和细菌)微生物群落和真核(真菌)微生物群落中的相对丰度和其绝对含量可得,如下:
taa+b=aauiss/raa+b_uiss
taf=aauiss/raf_uiss
获得绝对总量数据后,再根据古菌和细菌微生物群落各分类水平下(界、门、纲、目、科、属)各分类单元相对丰度组成(raa+b_taxon)及真菌微生物群落its基因各分类水平下(界、门、纲、目、科、属)各分类单元相对丰度(raf_taxon)组成计算其绝对含量:
aaa+b_taxon=taa+b×raa+b_taxon
aaf_taxon=taf×raf_taxon
至此,我们便获得了土样a和土样b中古菌、细菌、真菌各分类单元的绝对含量,可用于单样本跨域的比较。
通过实施例2的方法,结合三域内标菌株的绝对含量及其在原核和真核微生物群落结构中的相对丰度,计算出土壤样品a中古菌和细菌的绝对含量=(6.56×106±1.77×106copiesg-1)/(4.63±1.44%)-(6.56×106±1.77×106copiesg-1)copiesg-1=1.36×108±7.70×106copiesg-1、真菌的绝对含量=(6.56×106±1.77×106copiesg-1)/(10.86±4.58%)-(6.56×106±1.77×106copiesg-1)=6.32×107±3.39×107copiesg-1;土壤样品b中古菌和细菌的绝对含量=(8.24×106±5.21×105copiesg-1)/(0.95±0.15%)-(8.24×106±5.21×105copiesg-1)copiesg-1=8.82×108±2.02×108copiesg-1、真菌的绝对含量=(8.24×106±5.21×105copiesg-1)/(3.15±0.21%)-(8.24×106±5.21×105copiesg-1)=2.53×108±9.45×106copiesg-1(图3)。
此时,我们可采用绝对含量来比较土样a中子囊菌门和土样b中变形菌门微生物数量多少的差异,根据公式,土样a中子囊菌门绝对含量=(6.56×106±1.77×106copiesg-1)/(10.86±4.58%)×(73.20±6.38%)=5.26×107±2.94×107copiesg-1;土样b中变形菌门绝对含量=(6.56×106±1.77×106copiesg-1)/(4.63±1.44%)×(38.09±1.02%)=3.38×108±7.45×107copiesg-1)。传统的高通量测序结果,只能割裂地表征两个菌门在各域中的优势程度,我们可知子囊菌门(73.20±6.38%)在真菌中的优势程度显著高于变形菌门(38.09±1.02%)在细菌域中的优势程度,两个菌门间微生物的数量无法比较。而通过本发明中三域高通量绝对定量方法的应用,可对两个菌门的绝对含量进行跨样本跨域比较,我们得知虽子囊菌门在土样a的真菌域中优势程度更强,但其绝对含量(5.26×107±2.94×107copiesg-1)却显著少于土样b的细菌域中变形菌门(3.38×108±7.45×107copiesg-1),如图4所示。
对比例2、无三域通用内标菌株添加两个土壤样品中三域微生物垮样本跨域的比较
不添加三域通用内标菌株,直接对土壤样品a和b进行dna提取,并分别同时对两个样品的古菌域和细菌域、真菌域进行高通量测序,相关实验条件如测序条件、分析方法等均与实施例2保持一致。
所得结果为:因缺失三域通用内标菌株的添加,无法结合其相对丰度和绝对含量计算土壤样品a和b中古菌和细菌及真菌的绝对总量,从而无法进一步计算出土壤样品a中子囊菌门的绝对含量和土壤样品b中变形菌门的含量,两个菌门间微生物数量差异不得而知,进而无法跨样本跨域对两个菌门的绝对含量进行比较,微生物间的相互比较仅限于单样品单域内及其优势程度,进一步限制了多个样本间土壤三域微生物的相互比较及统筹分析。
相关参考文献:
batesst,berg-lyonsd,caporasojg,walterswa,knightr,fierern.examiningtheglobaldistributionofdominantarchaealpopulationsinsoil.theismejournal5,908-917(2011)[batesst,berg-lyonsd,caporasojg,walterswa,knightr,fierern.全球土壤中优势古菌种群分布研究.《isme杂志》5,908-917(2011)];
tedersool,bahramm,polmes,koljalgu,yorouns,wijesunderar,etal.globaldiversityandgeographyofsoilfungi.science346,1256688(2014)[tedersool,bahramm,polmes,koljalgu,yorouns,wijesunderar,等.全球土壤真菌的多样性和地理分布.《科学》346,1256688(2014)];
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
序列表
<110>浙江大学
<120>土壤三域微生物高通量绝对定量方法
<160>2
<170>siposequencelisting1.0
<210>1
<211>18
<212>dna
<213>人工序列(artificialsequence)
<400>1
gcggtaaggtgaagagtg18
<210>2
<211>19
<212>dna
<213>人工序列(artificialsequence)
<400>2
ctaacgaggctaacgagac19
- 还没有人留言评论。精彩留言会获得点赞!