一种大鼠长链非编码lncRNA-lncMSTRG10078及其抵抗细胞损伤的应用的制作方法
本发明涉及分子生物学技术领域,特别是涉及一种大鼠长链非编码lncrna-lncmstrg10078及其抵抗细胞损伤的应用。
背景技术:
lncrna是一类转录本长度超过200nt的rna分子,其缺乏特异完整的开放阅读框,不具有编码蛋白质的能力。根据lncrna在基因组中的相对位置将其分为正义lncrna、反义lncrna、双向lncrna、基因间lncrna、基因内lncrna5类,这种位置关系对于推测lncrna的生物学功能具有很大的帮助。根据功能将lncrna分为信号分子、诱饵分子、引导分子和骨架分子等4类分子。这些lncrna曾被认为是进化过程中累积的“垃圾序列”,是rna聚合酶ⅱ转录的副产物,不具有生物学功能,因而没有给予足够的重视。随着测序技术的不断发展,人们逐渐解开了lncrna的神秘面纱,越来越多的证据表明lncrna是一个具有多种功能的转录本。近年来的研究表明,lncrna广泛参与到细胞分化发育、染色体沉默、基因组印记、染色质修饰、转录激活、转录干扰等多种重要的生物学过程。
细胞损伤主要体现在炎症、凋亡、代谢改变以及线粒体功能损伤等方面。ros是细胞代谢的产物,作用于线粒体,会引起线粒体的损伤;由于细胞不能及时代谢处理ros,导致ros在细胞内大量积聚,引起细胞的功能紊乱,造成细胞损伤,最终导致细胞凋亡和cyp450代谢酶的改变。然而与编码蛋白质的mrna相比,lncrna的表达丰度很低,仍有大量的lncrna未被证实和发现。因此,研究发现并鉴定新型的lncrna,并研究其在抵抗细胞损伤方面的研究具有重要的意义。
技术实现要素:
针对上述现有技术,本发明的目的是提供一个大鼠长链非编码rna-lncmstrg10078及其在抵抗细胞损伤中的作用机制,进而应用该长链非编码rna抵抗细胞损伤。
为实现上述目的,本发明提供了如下方案:
作为本发明的第一方面:
本发明提供一种大鼠长链非编码lncrna,所述lncrna为lncmstrg10078,具有如seqidno.1所示的序列。
进一步地,所述大鼠长链非编码lncrna序列还包括在seqidno.1所示的序列中添加、取代、插入或缺失一个或多个核苷酸而生成的突变体、等位基因或衍生物。
作为本发明的第二方面:
本发明提供一种真核过表达载体,含有上述的大鼠长链非编码lncrna。
作为本发明的第三方面:
本发明提供上述的大鼠长链非编码lncrna在抵抗细胞损伤中的应用。
以及,上述的真核过表达载体在在抵抗细胞损伤中的应用。
作为本发明的第四方面:
本发明提供一种抵抗细胞损伤的方法,包括以下步骤:
(1)构建上述的大鼠长链非编码rna的真核过表达载体;
(2)将真核过表达载体转染至细胞中,使大鼠长链非编码rna的表达量增加。
本发明公开了以下技术效果:
本发明首次从大鼠中分离得到一个与抵抗细胞损伤相关的大鼠长链非编码rnalnc-mstrg10078。本发明的lnc-mstrg10078可作为基因资源或者开发成核酸药物在农业生产、畜禽养殖、人疾病治疗中使用,用于抵抗外源性药物或者毒物对机体的损伤作用,具有广泛的应用价值。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为lncrna-lncmstrg10078在大鼠基因组位置以及结构图;
图2为lncrna-lncmstrg10078编码能力的分析结果图,a为ncbiorf-finder预测结果,b为cpc网站预测结果;
图3为克隆lncrna-lncmstrg10078的扩增结果图,m表示dnamarker,dna分子量标准为dl5000,1-4泳道表示lncmstrg10078的扩增条带,大小为1964bp;
图4为quickcuthindiii和quickcutxhoi双酶切鉴定过表达载体pcdna3.0-lncmstrg10078,过表达载体的质粒大小为7334bp,所用dnamarker为dl15000bp;
图5为pcdna3.0-egfp转染条件摸索图;
图6为lncmstrg10078过表达验证结果图,结果表示为mean±sd(n=3),*表示p<0.05,与组内相比差异显著;**表示p<0.01,与组内相比差异极显著;
图7为lncmstrg10078调控线粒体相关基因表达图,结果表示为mean±sd(n=3),*表示p<0.05,与组内相比差异显著;**表示p<0.01,与组内相比差异极显著;
图8为lncmstrg10078调控炎症相关基因表达图,结果表示为mean±sd(n=3),*表示p<0.05,与组内相比差异显著;**表示p<0.01,与组内相比差异极显著;
图9为lncmstrg10078调控凋亡相关基因表达图,结果表示为mean±sd(n=3),*表示p<0.05,与组内相比差异显著;**表示p<0.01,与组内相比差异极显著;
图10为lncmstrg10078调控代谢相关基因表达图,结果表示为mean±sd(n=3),*表示p<0.05,与组内相比差异显著;**表示p<0.01,与组内相比差异极显著;
图11为lncmstrg10078对ros调控作用图;
图12为lncmstrg10078对细胞凋亡的调控作用图。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对本申请提供进一步的说明,本文使用的所有技术和科学术语具有于本申请所属技术领域的普通技术人员通常理解的相同含义。
正如背景部分所介绍,lncrna参与机体的多种生物学功能,但是关于细胞损伤相关的lncrna相比于mrna的研究尚且不足。因此,本发明的目的是提供一种大鼠细胞损伤相关的长链非编码rna,并将其应用于抵抗细胞的损伤。
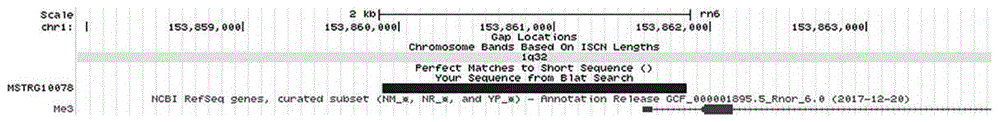
本发明利用cdna末端快速扩增技术(race)获得长链非编码rnalncrna-lncmstrg10078。通过ucsc和ncbi进行比对分析显示lncrna-lncmstrg10078定位于大鼠基因组1号染色体,长度为1964bp,其核苷酸序列如seqidno.1所示,结构如图1所示。
由于核苷酸序列的特殊性,任何seqidno.1所示多核苷酸的变体,只要其与该核苷酸具有90%以上同源性,且具有相同的功能,均属于本发明保护的范围之内。多核苷酸的变体是指一种具有一个或者多个核苷酸改变的多核苷酸序列,此核苷酸变体包括取代变异体、缺失变异体和插入变异体。
术语“同源”是主要是指序列上的同源,也就是用来说明两个或多个rna或dna序列具有相同的祖先。同源的序列一般有相似的功能。rna和dna的同源性常常通过它们序列的相似性来判定,相似性是指序列比对过程中用来描述检测序列和目标序列之间相同dna碱基或氨基酸残基顺序所占比例的高低。一般来说,当相似程度高于50%时,常推测检测序列和目标序列可能是同源序列;当相似性程度低于20%时,就难以确定其是否具有同源性。
通过编码能力预测网站分析lncmstrg10078的编码能力,显示lncmstrg10078不具有编码蛋白能力。
本发明还应用同源重组方法克隆lncmstrg10078序列,构建过表达载体,通过hindiii和xhoi酶切过表达载体pcdna3.0,胶回收线性化的过表达载体pcdna3.0和目的片段进行连接,测序鉴定,构建真核表达载体。用脂质体转染试剂,分别转染空载体pcdna3.0和目的载体pcdna3.0-lncmstrg10078到gh3细胞中,荧光定量pcr分析结果lncmstrg10078相关靶基因的表达量,结果显示,相比于转染空载体组,转染目的载体pcdna3.0-lncmstrg10078能够显著的抵抗细胞损伤作用。
用脂质体转染试剂,分别转染空载体pcdna3.0和目的载体pcdna3.0-lncmstrg10078到gh3细胞中用流式细胞仪检测ros以及细胞凋亡的发生,结果显示相比于转染空载体组,转染目的载体pcdna3.0-lncmstrg10078能够显著的降低ros以及细胞凋亡的发生,进而抵抗细胞损伤作用。
综上所述,利用真核过表达载体,使大鼠lncrna-lncmstrg10078增加表达量,可以显著的抵抗细胞的损伤作用。
为了使得本领域技术人员能够更加清楚地了解本申请的技术方案,以下将结合具体的实施例对本申请进行详细的说明。
本发明实施例中所用的试验材料均为本领域常规的试验材料,均可通过商业渠道购买得到。未注明详细条件的实验方法是按照常规试验方法或按照供应商所建议的操作说明书进行的。
实施例1lncrna-lncmstrg10078全长序列扩增
1.材料与试剂
1.1材料
本试验所使用的细胞系是大鼠垂体瘤细胞(gh3细胞)。
1.2试剂
phantamaxsuper-fidelitydnapolymerase,货号p515,购自南京诺唯赞生物科技有限公司;hiscriptii1ststrandcdnasynthesiskit(+gdnawiper),货号r212-01,购自南京诺唯赞生物科技有限公司;fastpuregeldnaextractionminikit,货号dc301,购自南京诺唯赞生物科技有限公司;mightyta-cloningreagentsetfor
2.实验方法
2.1总rna的提取
严格按照生工生物工程(上海)股份有限公司的柱式动物组织总rna抽提纯化试剂盒(b518651)从每个gh3细胞样本中分离总rna。用quawellq3000测定所提总rna样品的od260和od280,计算rna浓度。取1μg所提总rna样本用1.0%琼脂糖凝胶电泳方法检测总rna完整度。
2.2引物设计
根据已知的部分lncrna-lncmstrg10078的核苷酸序列,参考南京金斯瑞有限公司的引物计算工具设计引物,引物序列如下:
2.3反转录pcr
本次试验采用的反转录引物为qt和gsp-rt,根据引物合成的订单对引物进行稀释,每20μl反转录体系使用1μl,进行3’race和5’race的cdna的合成,完全按照诺唯赞hiscriptii1ststrandcdnasynthesiskit合成80μlcdna。
rnaseh处理:dna-rna杂合链中去除rna,在冰浴上向已经合成第一条链的每20μl反应体系中依次加入reactionbuffer(10x)2μl,depc水17.8μl,rnaseh(5u/μl)0.2μl,轻轻混匀(可以用移液器吹打混匀或用vortex在最低速度轻轻混匀),随后离心沉淀液体。37℃孵育1h,加入2.5μl0.5medta(ph8.0)混匀,以终止反应,后续使用乙醇沉淀法纯化合成的双链cdna。
说明:其他情况dna-rna杂合链中去除rna的条件可以参考上述条件进行。rnaseh反应的ph范围约在7.5-8.3均可。
2.43’race和5’race
3’race和5’race参考pcr技术实验指南(原著第2版)中的经典race法进行的。
2.5lncrna-lncmstrg10078结构和编码能力的分析
应用ucsc和ncbi网站对lncrna-lncmstrg10078的全长序列进行结构的分析,并应用ncbiorf-finder和cpc网站对lncrna-lncmstrg10078编码能力进行分析。
3.实验结果
应用5’和3’race技术对lncrna-lncmstrg10078的全长进行鉴定,最终获得其全长序列为1964bp,如seqidno.1所示。应用ucsc和ncbi进行序列的比对,其结果显示lncrna-lncmstrg10078主要位于大鼠的1号染色体(反义链,从153859895-153861846)(图1)。应用ncbiorf-finder和cpc,图2中可以得出,lncrna-lncmstrg10078不具有蛋白编码能力,上述结果证明了lncrna-lncmstrg10078是一个全长为1964bp且不具有蛋白编码能力的lncrna。实施例2lncrna-lncmstrg10078序列的克隆与分析
1.试剂与载体
phantamaxsuper-fidelitydnapolymerase,货号p505,购自南京诺唯赞生物科技有限公司;hiscriptii1ststrandcdnasynthesiskit(+gdnawiper),货号r212-01,购自南京诺唯赞生物科技有限公司;fastpuregeldnaextractionminikit,货号dc301,购自南京诺唯赞生物科技有限公司;clonexpressultraonestepcloningkit,货号c115,购自南京诺唯赞生物科技有限公司;mightyta-cloningreagentsetfor
2.方法
2.1总rna的提取
实验方法同实施例1中2.1总rna提取。
2.2引物设计
根据lncrna-lncmstrg10078核苷酸的序列,如seqidno.1所示,应用同源重组酶的设计原则,设计lncrna-lncmstrg10078扩增引物,具体如下:
lncmstfactatagggagacccaagcttttcccaatggattcaaggagc;lncmstrccctctagatgcatgctcgagtttttttttttttgactgtttcaatcttttaaacaagatt
2.3第一链cdna合成反应
反转录反应分为三步,以提取的总rna(<5μg)为模板,首先进行rna模板变性,在rnase-free离心管中配制反应液oligo(dt)23vn(50μm),添加rna和rnase-freeddh2o后至体积12μl,将上述反应液,65℃加热5min,迅速置于冰上骤冷,并在冰上静置2min。将上述反应液变性后,继续添加4×gdnawipermix4μl,用移液器轻轻吹打混匀。42℃2min,去除基因组dna。继续在上一步的混合液中,继续添加10×rtmix2μl、hiscriptiienzymemix2μl,用移液器轻轻吹打混匀后,50℃45min,85℃2min进行反转录反应得到第一链cdna。
2.4lncrna-lncmstrg10078转录本pcr扩增
使用高保真酶phantamaxsuper-fidelitydnapolymerase试剂盒在pcr仪上进行pcr反应,反应体系如下:2×phantamaxbuffer25μl,dntpmix(10mmeach)1μl,lncmstrg10078特异性上下游引物各2μl,模板dna2μl,phantamaxsuper-fidelitydnapolymerase1μl,ddh2o至50μl。反应程序为预变性95℃3min;变性95℃15sec,退火55℃15sec,延伸72℃120sec,循环35次;然后72℃延伸5min。获得的pcr产物进行琼脂糖电泳检测,使用凝胶成像仪拍照,分析所得的片段大小(图3)并切取正确的单一目的条带。
2.5lncrna-lncmstrg10078过表达载体构建
将过表达载体pcdna3.0用quickcuthindiii和quickcutxhoi进行双酶切,琼脂糖凝胶电泳后切取正确单一条带。将2.4中的目的基因条带与过表达载体双酶切的条带进行胶回收,胶回收步骤按照omega的胶回收试剂盒进行。胶回收的dna溶液用2×cloneexpressmix进行连接,连接体系如下:线性化载体3μl、lncmstrg10078目的片段2μl、2×cloneexpressmix5μl,使用移液器轻轻吹打混匀,短暂离心将反应液收集至官底,将混合液置于pcr仪中50℃15min,降至4℃或立即置于冰上冷却。将连接产物转化至trans-t1phageresistant化学感受态细胞,取50μl冰浴上融化的感受态细胞,加入5μl连接产物,轻轻混匀,在冰浴中放置30分钟,42℃水浴热激30秒,然后快速将管转移到冰浴中2分钟,该过程不要摇动离心管。向每个离心管中加入500μl无菌的不含抗生素的lb培养基,混匀后置于37℃,200rpm培养1小时,使细菌复苏。5000rpm离心1min后,弃去上清至剩余100μl,重悬细胞沉淀加入到含有氨苄抗生素的lb琼脂培养基上,将细胞均匀涂开。将平板至于37℃至液体被吸收,倒置平板,37℃过夜培养,挑取单克隆摇菌12h,提取质粒后利用quickcuthindiii和quickcutxhoi进行双酶切鉴定(图4),将鉴定正确的单克隆载体送去生工生物工程(上海)股份有限公司测序鉴定。
3.实验结果
经琼脂糖电泳检测以及测序结果分析,所得的阳性克隆的片段长度为1964bp,且其序列正确,证实了lncrna-lncmstrg10078的真实存在。
实施例3:lncrna-lncmstrg10078功能验证
1.gh3细胞培养
gh3细胞复苏于含有5mldmem完全培养基(10%fbs+90%dmem+1ml双抗+1ml谷氨酰胺)中并置于95%相对湿度,温度37℃,并含有5%co2的培养箱中培养。当细胞生长状态达到70%~90%时,采用胰蛋白酶-edta消化法,按1:2比例传代,每1-2天传代一次。细胞冻存采用细胞冻存液(90%fbs+10%dmso)进行冻存。
2.gh3细胞转染条件摸索
转染前一天,胰酶消化细胞并计数(使其在大约24小时后转染前密度达到90%左右为宜),将细胞铺板在1ml含血清,不含抗生素的正常生长的培养基中;用50μl无血清的opti-mem培养基稀释1:1,1:1.5,1:2,1:2.5,1:3的exfecttransfectionreagent试剂,静置活化5min;对于每孔细胞,使用50μl无血清的opti-mem培养基稀释1μg质粒,轻轻混匀后于室温放置5min;将稀释好的exfecttransfectionreagent试剂和dna混合,轻柔混匀,室温放置20min,以便形成dna/exfecttransfectionreagent试剂混合物;在静置20min的期间处理细胞,然后加入900μlopti-mem;将exfecttransfectionreagent试剂混合物混合物加入到各个孔中,来回轻柔摇晃细胞培养板,做好标记;6h后更换成无抗完全培养基,并于转染后12小时、24小时、36小时和48小时记录荧光,确定最佳转染浓度和时间(图5)。
3.lncrna-lncmstrg10078转染
根据2确定最佳的转染浓度以及时间(1:2.5,转染24h)。转染前一天,胰酶消化细胞并计数(使其在大约24小时后转染前密度达到90%左右为宜),将细胞铺板在1ml含血清,不含抗生素的正常生长的培养基中;按照dna:转染试剂=1:2.5的比例,用50μl无血清的opti-mem培养基稀释的2.5μlexfecttransfectionreagent试剂,静置活化5min;对于每孔细胞,使用50μl无血清的opti-mem培养基稀释1μg质粒,轻轻混匀后于室温放置5min;将稀释好的exfecttransfectionreagent试剂和dna混合,轻柔混匀,室温放置20min,以便形成dna/exfecttransfectionreagent试剂混合物;在静置20min的期间处理细胞,然后加入900μlopti-mem;将exfecttransfectionreagent试剂混合物加入到各个孔中,来回轻柔摇晃细胞培养板,做好标记,并于6h后更换成无抗完全培养基,并于转染24h后进行收样。4.lncrna-lncmstrg10078相关功能基因的荧光定量pcr检测
4.1总rna提取
实验方法同实施例1中2.1总rna提取。
4.2引物设计
根据相关靶基因的核苷酸序列,通过ncbiprimerblast设计qrt-pcr引物,产物长度在70-300bp之间,使用表达量稳定的β-actin作为内参基因。引物均由南京金斯瑞生物科技有限公司合成。
引物序列如下:
4.3反转录反应
反转录反应分为两步,以提取的总rna1μg为模板,首先进行基因组dna去除,在rnase-free离心管中配制反应液oligo(dt)23vn(50μm)1μl,randomhexamers(50ng/μl)1μl,4×gdnawipermix4μl,添加rna和rnase-freeddh2o后至体积16μl,用移液器轻轻吹打混匀,42℃加热2min,迅速置于冰上骤冷,在上述反应液中继续添加10×rtmix2μl、hiscriptiienzymemix2μl,用移液器轻轻吹打混匀后,50℃15min,85℃2min进行反转录反应得到第一链cdna。
4.4quantitativereal-time(rt)pcr
使用chamquniversalsybrqpcrmastermix试剂盒在cfx96real-timepcrdetectionsystem实时荧光定量pcr仪上进行pcr扩增,在冰上配制qrt-pcr反应体系如下:
反应混合物轻微离心后,于cfx96real-timepcrdetectionsystem实时荧光定量pcr仪上进行pcr扩增,反应程序为:
第一步:30sat95℃;
第二步:10sat95℃;30sat对应的tm值,共40个循环;
第三步:10sat95℃;65℃to95℃,递增0.5℃。分析各基因的扩增曲线和溶解曲线。通过ct比较法,并以β-actin作为内参基因,即2-δδct来测算转录水平差异。实验重复3次。
5.实验结果
过表达pcdna3.0及pcdna3.0-lncmstrg10078的gh3细胞中lncmstrg10078的表达情况见图6,由此可以看出lncmstrg10078过表达成功。从图7、图8、图9、图10可以看出,lncmstrg10078能够显著的抑制调控线粒体呼吸链相关亚基变化、显著的抑制炎症发生、显著的抑制凋亡的发生、显著的调控线粒体代谢相关的cyp450酶变化。实验结果表明,lncmstrg10078在过表达pcdna3.0及pcdna3.0-lncmstrg10078的gh3细胞中具有重要的作用,能够调控线粒体呼吸链亚基、炎症和凋亡的发生、以及代谢的改变的角度,进一步的抵抗细胞损伤作用。
实施例4lncrna-lncmstrg10078对ros以及细胞凋亡的调控
1.gh3细胞培养
gh3细胞培养条件同实施例3中的1。
2.lncrna-lncmstrg10078转染
lncrna-lncmstrg10078转染同实施例3中的3。
3.ros测定
gh3细胞培养后,接种于12孔板,分别转染pcdna3.0和pcdna3.0-lncmstrg1007824h后,加入含有10μmdcfh-da的培养基。37℃孵育细胞1h。用0.25%胰酶消化细胞30s,加入完全培养基终止消化,制成细胞悬液,1000g离心5分钟,用pbs洗涤1~2次,离心收集细胞沉淀物,用500μlpbs重悬细胞,用beckmanfc500进行ros测定。
4.细胞凋亡测定
gh3细胞培养后,接种于12孔板,分别转染pcdna3.0和pcdna3.0-lncmstrg1007824h后,用不含edta的0.25%胰酶消化细胞30s,加入完全培养基终止消化,制成细胞悬液,在室温中2000rpm离心5分钟,用预冷pbs重悬细胞一次,2000rpm离心5分钟,弃上清洗涤细胞,加入300μl的1×bindingbuffer悬浮细胞,再加入5μl的annexinv-fitc混匀后,避光,室温孵育15分钟;上机前5分钟再加入5μl的pi染色,上机前补加200μl的1×bindingbuffer,用beckmanfc500进行细胞凋亡的测定。
5.实验结果
从流式结果图11和图12可以得出,gh3细胞中过表达pcdna3.0及pcdna3.0-lncmstrg10078,lncmstrg10078能够显著的抑制ros的产生以及显著的抑制细胞的凋亡率,进一步说明lncmstrg10078在抵抗细胞的损伤中具有重要的作用与功能。
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
序列表
<110>华中农业大学
<120>一种大鼠长链非编码lncrna-lncmstrg10078及其抵抗细胞损伤的应用
<160>1
<170>siposequencelisting1.0
<210>1
<211>1964
<212>dna
<213>人工序列(artificialsequence)
<400>1
ttcccaatggattcaaggagctagcccccgcccctcgtccgaggccacctgctccgtgct60
tggttgcgatgttcacccccgcagccaacaaagcgaaagcaagcccagaaagcagatccc120
gtgagggtgacagcctgtgctggcctccatacagagaatactctccgaggggtccctgca180
ccctcagtacctgaggttggccttgtcttctccacttacctgcctaactggagaagctca240
agtctgtgagaagctcaagtctgtgcagccccactgcaggcaggtagggaaagacgctac300
tgtcccctgtcacactctggccaaaggcagcaccgcaccactcaggcagtacgagggact360
gctgtgttcagtaaaatccttgccttgcaggaccagacttcccttgaggatacgcgtggg420
ggctcttcgtatcaagatgtaacaagatgaagctcactttcctcccacatgagggctttt480
gtgcttttgtaccttggcactctctatccagaggacttgaggatcatcatgctccctgcc540
tcttgagcaaggctctgagcaaggatgctgtcctgtgaagtaaccattagccacatgtag600
ctgtaatactctaaataaaatacaatttaaaatccatttccccagtcccagcaagcagtt660
ctttttaaagcagttgggagcatgtgtgagtgcatataggtacagaacacttccaacttt720
gcagaacgttctcttgtcttgatctgagtctcccacttcctagggaggtttctaagaacc780
ccatgaagccagtcatagatgttagggtcacaccttaatccctgaatttaaatgtatact840
tcctctcagtattcaagcgattccttctgaaggaaatacatctcactgtgtatgggataa900
cacccaagtcgtcaggaggccttggttaaccccagcctggttcctgacaaggtctaatgc960
ttgtcctacttcacactgtgctgtccttgaggtctcccctcacttgcttctccccatttc1020
tccaccttacttccctgcctccacctcggggcttttgtatctgctaatgcttttgcatgg1080
atcctcacactccaatcttccaaacaactgccacatctcatctctgaggtggcccctaat1140
atgtcaacttgggggcctttattgtaggtattcccttcagtctgaccatcactaggggga1200
acataggaaaaaggtatcccctatgtgtaaacgaacaagtcagttaccagctcacagctc1260
ttgtgctttattctctgcctcccccttacactacaggcttcactgggcttgggtctttac1320
ctgcaaatccatttgtcctcagggcttgacgtgtctttgtacaataaaaacatgtttggt1380
gagggaaggtgagtggcttccctgttgatcaggactatgagctctcagaatggctgccag1440
acagatggtgtggttctctcagactcactttcaaaccgcatcccattctctggtcccaac1500
tgcaaggcaggcataatggtactgtcagggtcctcctgcctggcctcaggattccatctc1560
ctcggaagtgatgggaattagtgccttcccctgaagcctctgtcctttctgctccaattt1620
ctagttctcagctagaatgatctatattcaattgtactggataaaagaatgtcgattctc1680
tttagatgatatttggtatataggtcacacttcagagcaggccccatgccctggactaat1740
tagctatctgggcaacacagactgaattccatggggtttgtttgtttgtttgttcatttg1800
ttttgagtcagagggcaaacataaagttggatgggtaggaaggaggggaggacctgggaa1860
cagctgggagaacaagatgaacatgatcgattatgttttatgaaacctccaatgaataaa1920
taaaaatcttgtttaaaagattgaaacagtcaaaaaaaaaaaaa1964
- 还没有人留言评论。精彩留言会获得点赞!