一种二丙酸咪唑苯脲杂质A及其杂质A制备方法与流程
本发明涉及一种二丙酸咪唑苯脲杂质A及其杂质A制备方法,属于医药技术领域。
背景技术:
二丙酸咪唑苯脲,化学名为N,N'-二[3-(4,5-双氢-1H-咪唑-2-基)苯基]脲·二丙酸盐。1969年Schmidt等在应用鼠骆巴贝西虫筛选均二苯脲类化合物时,发现该药具有极好的抗梨形虫作用。同年Beveridge等进一步证实该药的ED50低于其它通用的抗梨形虫药物,二丙酸咪唑苯脲对马驽巴贝西虫、马巴贝西虫、牛双芽巴贝西虫、牛巴贝西虫、阿根廷巴贝西虫、分离巴贝西虫和犬巴贝西虫等各种巴贝西虫均有良好的治疗作用。由于该药具有能在体内长效的特性及发现该药尚可抑制梨形虫传播者-蜱体内虫体的感染力,因此家畜梨形虫的预防和治疗的药物首选咪唑苯脲。该药已广泛应用于世界各地,为国际公认的最佳抗梨形虫药物。在市场上由于其优良的药效,较低的成本,其逐步取代了三氮脒,成为抗梨形虫病的主打药物。其化学结构式如下:
在药物API研究过程中,杂质的研究是整个研究的关键,二丙酸咪唑苯脲为注射用原料,其含有的杂质,将影响产品使用的安全性。通过对二丙酸咪唑苯脲的稳定性研究以及制备工艺的深入研究,发现产品中有一个比较难以清除的杂质,且该杂质也是主要的降解杂质。因此,对二丙酸咪唑苯脲中的杂质相关研究意义重大,为二丙酸咪唑苯脲安全用药提供重要的指导意义,现有技术中,已经公开了多种二丙酸咪唑苯脲的制备方法以及提纯方法,但是现有文献中,未见对二丙酸咪唑苯脲杂质的相关报道。
技术实现要素:
针对现有技术的不足,本发明提供一种二丙酸咪唑苯脲杂质A及其杂质A制备方法。
一种二丙酸咪唑苯脲杂质A,所述的二丙酸咪唑苯脲杂质A结构式如下式Ⅰ所示:
根据本发明优选的,所述的二丙酸咪唑苯脲杂质A以其碱式和/或以其盐的形式存在。
进一步优选的,所述盐为:丙酸盐、乙酸盐、盐酸盐或者能够与碱式二丙酸咪唑苯脲杂质A成盐中的一种或两种以上混合。
本发明提供了二丙酸咪唑苯脲杂质A的制备方法。
上述二丙酸咪唑苯脲杂质A的制备方法,按如下方法a、b中的其中一种进行:
方法a包括步骤如下:
(1)将2-(3-硝基苯基)咪唑啉加入到有机溶剂和水的混合溶液中,得混合液,搅拌状态下向混合液中加入碱反应,反应完成后,析出固体、抽滤得到中间体1;
(2)将步骤(1)得到的中间体1溶解在溶剂a中,加入Pb/C,控制反应温度为30-50℃,开始通入氢气,控制反应压力,检测反应完毕,停止反应,过滤除去Pb/C,浓缩溶剂,析晶,得中间体2;
(3)将中间体2溶解在溶剂b中,加入三光气、碱,搅拌反应,反应完成后向体系中缓慢滴加2-(3-氨基苯基)咪唑啉溶液,滴加完成后反应一段时间后进行分离纯化,得到杂质A;
方法b包括步骤如下:
将咪唑苯脲溶解在有机溶剂与水的混合溶液中,加入碱,在35-45℃下反应,控制温度、碱量以及反应时间,然后用洗脱剂进行柱分离,分离得到杂质A。
根据本发明优选的:
方法a,步骤(1)中所述的有机溶剂为醇类、乙腈、丙酮或者四氢呋喃中的一种或者两种以上以任意比混合。
优选的,所述的有机溶剂为醇类,进一步优选的,所述的有机溶剂为甲醇。
方法a,步骤(1)中,混合液中2-(3-硝基苯基)咪唑啉:甲醇:水的质量比为1:(5-15):(0.1-5)。
方法a,步骤(1)中,所述的碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾或者有机碱,碱的加入量为2-(3-硝基苯基)咪唑啉摩尔质量的0.5-5倍。
进一步优选的,所述的碱选自氢氧化钠。
方法a,步骤(1)中,反应温度为25-50℃,反应时间为2-12h。
方法a,步骤(2)中所述的溶剂a为醇类、酯类或酮类溶剂,溶剂a的加入量为中间体1质量的3-30倍。
进一步优选的,所述溶剂a为醇类溶剂,所述醇类溶剂为甲醇。
方法a,步骤(2)中Pb/C加入量为中间体1质量的2-50%,反应压力为0.2-0.4Mpa,反应时间为20-60min。
进一步优选的,Pb/C加入量为中间体1质量的5-10%。
方法a,步骤(3)中所述的溶剂b为丙酮、四氢呋喃、甲苯、乙酸乙酯、1,4-二氧六环中一种或者两种以上以任意比混合;所述的碱为三乙胺、吡啶或N,N-二异丙基乙胺;溶剂b的加入量为中间体2质量的3-10倍,碱的加入量为中间体2摩尔质量的0.3-3倍,加入三光气的量为中间体2摩尔质量的0.35-2.5倍,反应时间为1-3h。
方法a,步骤(3)中2-(3-氨基苯基)咪唑啉溶液中2-(3-氨基苯基)咪唑啉为中间体2摩尔量的0.5-2倍,溶解2-(3-氨基苯基)咪唑啉的溶剂为甲苯,甲苯用量为2-(3-氨基苯基)咪唑啉质量的2-5倍。
进一步优选的,加入三光气的量为中间体2摩尔质量的1-1.5倍,反应时间为1.5h。
进一步优选的,2-(3-氨基苯基)咪唑啉溶液中2-(3-氨基苯基)咪唑啉为中间体2摩尔量的1-1.2倍。
方法b中,所用有机溶剂与方法a步骤(1)中的有机溶剂相同,有机溶剂的用量为咪唑苯脲质量的5-15倍,水的用量为咪唑苯脲质量的0.1-5倍,所述的碱与方法a步骤(1)中的碱相同;碱的用量为咪唑苯脲摩尔质量的0.5-5倍,反应时间为2-6h。
方法b中,所述的洗脱剂为乙酸乙酯与石油醚的混合物或者甲醇与二氯甲烷的混合物。
优选的,所述的洗脱剂为乙酸乙酯与石油醚的混合物,乙酸乙酯:石油醚的质量比为1:1-5。
本发明的合成路线:
方法a的合成路线如下:
方法b的合成路线如下:
然后通过柱分离对两个杂质进行分离纯化,得到杂质A。
本发明的有益效果:
1、本发明的二丙酸咪唑苯脲杂质A对二丙酸咪唑苯脲质量的提高,以及从源头上控制二丙酸咪唑苯脲的安全性具有重要的现实意义。
2、本发明的二丙酸咪唑苯脲杂质A可用于二丙酸咪唑苯脲原料药和制剂的注册、质量研究、工艺研究、杂质制备和作为杂质对照品,对制定和提高二丙酸咪唑苯脲的质量标准提供有效的数据支持,为二丙酸咪唑苯脲的临床安全使用提供有效保障。
附图说明
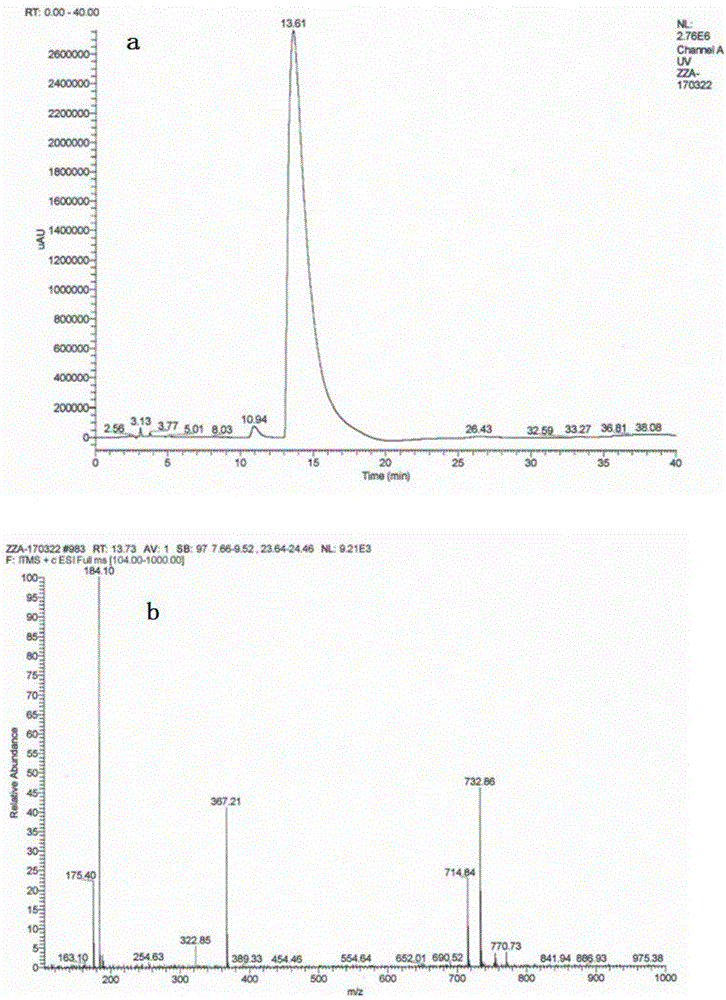
图1为实施例1所得的杂质A的HPLC-MS色谱图及MS色谱图,其中a为HPLC-MS色谱图,b为MS色谱图。
图2为实施例1所得的杂质A的核磁共振谱图,其中图2-1是杂质A在DMSO中的1H-NMR图普,图2-2是杂质A在DMSO中的1H-NMR部分放大图普,图2-3是杂质A在CD3OD中的1H-NMR图普,图2-4是杂质A在CD3OD中的1H-NMR部分放大图普,图2-5是杂质A的13C-NMR图.
图3为实施例1所得的杂质A的红外光谱图。
具体实施方式:
为了进一步阐明本发明,下面给出一系列的实施例,这些实施例完全是例证性的,它们仅用来对本发明具体描述,不应当理解为对本发明的限制。利用本发明所述的技术方案,或本领域的技术人员在本发明技术方案的启发下,设计出类似的技术方案,而达到相似的技术效果的,均是落入本发明的保护范围。
实施例1
二丙酸咪唑苯脲杂质A的制备方法,步骤如下:
1)中间体2的制备
称取2-(3-硝基苯基)咪唑啉15g,加入甲醇100ml,水30ml,氢氧化钠3.2g,升温至35℃反应5小时,反应完毕,浓缩出甲醇,用稀盐酸调节pH值为2.5-3.5,析出固体,抽滤得中间体1,约13.5g。
取中间体1加入100ml甲醇中溶解,然后加入Pb/C 1.0g,然后通氢气,控制反应温度为40℃,开始加氢,控制压力为0.3Mpa,观察压力表,若压力稳定不变认为反应完毕,过滤,除去Pb/C,然后浓缩甲醇,加入水,析晶。得中间体2产品约10.5g,纯度98.2%。
2)杂质A的制备
取中间体2 10g,加入100ml甲苯完全溶解,然后加入三光气17.0g,-2℃下缓慢滴加三乙胺11.3g,滴加完毕后升温至室温反应30min,然后降温至-10℃,将2-(3-氨基苯基)咪唑啉9.0g溶解在30ml甲苯中,溶解完毕后缓慢滴加至中间体2与三光气的体系中,滴加过程中控制温度不超过-5℃。滴加完毕后,升温至0℃下继续反应1h,反应完毕后加入100ml水,调节pH至1.8-2.5,搅拌1h,过滤,滤饼加入四氢呋喃打浆,过滤,得杂质A14.2g,纯度95.2%。
采用液质连用技术(HPLC-MS),结合二丙酸咪唑苯脲的制备反应机理,对实施例1的二丙酸咪唑苯脲杂质A进行检测,
所述的液质连用技术色谱条件如下:
色谱柱:XBridge C18,250mm×4.6mm,5μm柱,柱温:35℃,检测波长:240nm,流速:1.0ml/min,进样量:20μl,流动相:甲醇:乙腈:乙酸铵缓冲液(7.5∶7.5∶85),乙酸铵缓冲液的制备:乙酸铵0.77g,溶于800ml水中,冰乙酸调节pH值为4.5±0.1,加水至1000ml,
测得的HPLC-MS色谱图如图1所示,通过图1可以看出,其出峰位置相对于咪唑苯脲相对保留时间为0.7,将上述杂质分别加入含有杂质的样品中,可以看出杂质相对保留时间为0.7处的杂质峰增大,杂质A相对保留时间约0.7,纯度为0.05%-0.5%左右;
HPLC-MS测定中,测得的杂质A的分子量为366;该杂质的分子量比主产品咪唑苯脲的分子量分别多18,推测是咪唑苯脲加水的化合物,结合二丙酸咪唑苯脲的结构特点,推测是二丙酸咪唑苯脲中官能团咪唑啉环双键打开,与水加成产物。
对杂质A进行核磁共振图谱以及红外图谱的测定,测得的核磁共振谱图及红外图谱如图2、图3所示,通过图2、图3均表明为杂质A的结构。
实施例2
二丙酸咪唑苯脲杂质A的制备方法,步骤如下:
1)中间体2的制备
称取2-(3-硝基苯基)咪唑啉15g,加入乙腈150ml,水100ml,碳酸钠8.3g,升温至40℃反应5小时,反应完毕,浓缩出甲醇,用稀盐酸调节pH值为2.5-3.5,析出固体,抽滤得中间体1,约15.1g。
取中间体1加入300ml乙酸乙酯中溶解,然后加入Pb/C 1.5g,然后通氢气,控制反应温度为30-50℃,开始加氢,控制压力为0.2-0.4Mpa,观察压力表,若压力稳定不变认为反应完毕,过滤,除去Pb/C,然后浓缩溶剂,加入水,析晶,得中间体2产品约11.6g,纯度97.6%。
2)杂质A的制备
取中间体2约10g,加入50ml乙酸乙酯完全溶解,然后加入三光气20g,-5~5℃下缓慢滴加三乙胺11.3g,滴加完毕后升温至室温反应30min,然后降温至-10℃,将2-(3-氨基苯基)咪唑啉9.0g溶解在30ml乙酸乙酯中,溶解完毕后缓慢滴加至中间体2与三光气的体系中,滴加过程中控制温度不超过-5℃。滴加完毕后,升温至0℃下继续反应1h,反应完毕后加入100ml水淬灭反应,然后浓缩,除去部分丙酮,调节pH至1.8-2.5,过滤。得杂质A 14.1g,纯度92.9%。
实施例3
二丙酸咪唑苯脲杂质A的制备方法,步骤如下:
取50g咪唑苯脲,加入500ml乙腈,100ml水,然后向溶液中加入氢氧化钾10g,升温,在40℃下搅拌反应,反应2-3h,原料大部分转化后结束反应,过滤,母液浓缩干,得到杂质A粗品,将该粗品经过柱层析提纯,洗脱剂为乙酸乙酯:石油醚=1:1,经过分离后得到杂质A约3g,纯度96.1%。
实施例4
二丙酸咪唑苯脲杂质A的制备方法,步骤如下:
取50g咪唑苯脲,加入500ml甲醇,200ml水,然后向溶液中加入氢氧化钾18g,升温,回流下搅拌反应,反应1-2h,原料大部分转化后结束反应,过滤,母液浓缩干,得到杂质A粗品,将该粗品经过柱层析提纯,洗脱剂为甲醇:二氯甲烷=1:8,经过分离后得到杂质A约2.7g,纯度95.2%。
- 还没有人留言评论。精彩留言会获得点赞!