Cas9过表达载体及其构建方法和应用与流程
cas9过表达载体及其构建方法和应用
技术领域
[0001]
本发明涉及基因编辑技术领域,具体涉及一种cas9过表达载体及其应用。
背景技术:
[0002]
crispr/cas9(clustered regularly interspaced short palindromic repeats)系统是继znf和talens技术之后的第三代基因编辑技术,被广泛应用于细胞水平和个体水平基因编辑细胞系和模式动物的建立。
[0003]
crispr/cas相比其它基因组编辑技术,如锌指核酸酶(zfns)或转录激活因子样效应物核酸酶(talens),操作更简单,同时更容易针对同一细胞中的多个基因位点进行编辑,该系统可成为替代目前基因组工程方法的一种更安全,毒性更低的新方法。
[0004]
到目前为止crispr/cas已经运用到了各种细胞以及大肠杆菌、酿酒酵母、拟南芥、烟草、高粱、水稻、小麦、线虫、果蝇、蚕、斑马鱼、非洲爪蟾、小鼠、大鼠、猪、羊等,从最低等的微生物到哺乳动物的基因敲除修饰或突变。另一方面,其强大的基因组编辑能力已经用于生物治疗,如hiv、hbv等rna病毒引起的疾病治疗研究、单基因或多基因遗传病治疗研究、癌症治疗研究等方面。
[0005]
px330-u6-chimeric_bb-cbh-hspcas9是来源于feng zhang lab的最早运用crispr/cas9基因编辑技术实现哺乳动物基因编辑的载体,经大量的实验验证,它可以有效地实现哺乳动物细胞的基因编辑。但其cas9蛋白的表达量及核定位能力较有限,并且没有荧光及抗性筛选标记。
技术实现要素:
[0006]
本发明提供一种可以提高基因编辑效率的cas9过表达载体,解决了传统cas9表达载体表达量及核定位能量较弱的问题。
[0007]
一种cas9过表达载体,由原始载体px330-u6-chimeric_bb-cbh-hspcas9改造而成,原始载体具有grna骨架序列、cmv增强子和cas9基因,其特征在于,所述cmv增强子的下游插入ef1a启动子以替换原始载体的chickenβ-actin启动子;所述cas9基因的n端及c端再各插入一个核定位编码序列nls,所述cas9基因的c端核定位编码序列nls的下游依次插入wpre序列、3’ltr序列和bgh polya序列。
[0008]
进一步地,所述cas9基因c端的核定位编码序列nls与wpre序列之间插入序列p2a-egfp-t2a-puro。
[0009]
进一步地,所述grna骨架序列替换为如seq id no:1所示序列,去除原始载体中的无效多余序列。
[0010]
本发明改造px330-u6-chimeric_bb-cbh-hspcas9载体的如下原件:
[0011]
(1)去除残留的grna骨架序列,降低干扰。
[0012]
(2)改造启动子:将原有启动子(chickenβ-actin promoter)改造为具更高表达活性的ef1a启动子,增加cas9基因的蛋白表达能力。
[0013]
(3)增加核定位信号:在cas9的n端及c端均增加核定位信号编码序列(nls),增加cas9的核定位能力。
[0014]
(4)增加双筛选标记:原载体无任何筛选标记,不利于阳性转化细胞的筛选和富集,在cas9的c端,插入p2a-egfp-t2a-puro,赋予载体荧光和抗性筛选能力。
[0015]
(5)插入wpre和3’ltr等序列:在基因读码框最后插入wpre、3’ltr等序列,可增强cas9基因的蛋白翻译能力。
[0016]
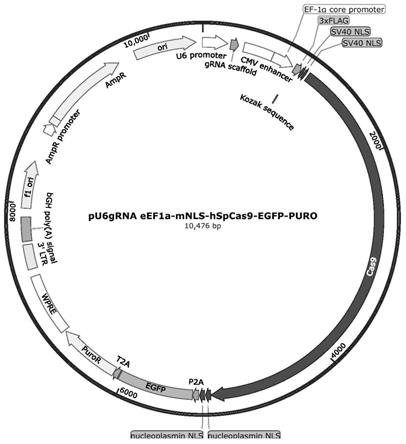
改造后得到的pu6grna eef1a-mnls-hspcas9-egfp-puro载体其碱基序列如seq id no:70所示,图谱如图13所示。
[0017]
本发明还提供一种cas9过表达载体的构建方法,它以载体px330-u6-chimeric_bb-cbh-hspcas9为原始载体,原始载体具有grna骨架序列、cmv增强子和cas9基因,其特征在于,包括以下步骤:(1)将所述cmv增强子下游的chickenβ-actin启动子替换为ef1a启动子;(2)在所述cas9基因的n端和c端各增加一个核定位编码序列nls,在所述cas9基因的c端核定位编码序列nls的下游依次插入wpre序列、3’ltr序列和bgh polya序列。
[0018]
进一步地,包括:将所述grna骨架序列替换为如seq id no:1所示序列,去除原载体中无效多余序列。
[0019]
进一步地,在所述cas9基因的c端nls与wpre序列之间插入p2a-egfp-t2a-puro序列。
[0020]
本发明的构建方法中包括对原始载体5个位点的改造,对各位点的改造顺序可以以任一顺序进行,更进一步优选地,所述构建方法按如下步骤进行:
[0021]
(1)将所述grna骨架序列改造为如seq id no:1所示序列;
[0022]
(2)将所述cmv增强子下游的chickenβ-actin启动子改造为ef1a启动子;
[0023]
(3)在所述cas9基因的n端增加至少一个核定位编码序列nls;
[0024]
(4)在所述cas9基因的c端依次插入至少一个核定位编码序列nls、p2a-egfp-t2a-puro序列、wpre序列、3’ltr序列和bgh polya序列。
[0025]
本发明的表达载体采用cmv增强子-ef1a启动子杂合启动子元件,增加cas9基因的蛋白表达能力,进一步地,同时在cas9基因n端及c端各增加数个核定位信号编码序列(nls),增加cas9的核定位能力。另外,本发明在载体中加入了荧光标记及抗性标记,使其更方便运用于载体阳性转化细胞的筛选及富集。
[0026]
本发明还提供了包含所述cas9过表达载体的crispr/cas9系统。
[0027]
本发明还提供了所述的crispr/cas9系统在构建突变型猪细胞系中的应用。
[0028]
本发明还提供了采用所述crispr/cas9系统构建突变型猪细胞系的方法,包括以下步骤:将所述crispr/cas9系统转入猪成纤维细胞,筛选突变型细胞株。
[0029]
与现有技术相比,本发明具有至少以下有益效果:
[0030]
本发明中经过实验验证经过改造的pu6grna eef1a-mnls-hspcas9-egfp-puro载体相对改造前的px330载体,更换了更强的启动子及添加了增强蛋白翻译的元件,提高了cas9的表达,并且增加了核定位信号个数,提高了cas9蛋白的核定位能力,具有更高的基因编辑效率。本发明在载体中加入了荧光标记及抗性标记,使其更方便运用于载体阳性转化细胞的筛选及富集。
附图说明
[0031]
图1为原始载体px330-u6-chimeric_bb-cbh-hspcas9的结构图谱。
[0032]
图2为原始载体经bbsi、xbai酶切电泳结果图。
[0033]
图3为全基因合成的grnasc1-6插入片段电泳图谱。
[0034]
图4为重组载体pu6grnacas9载体的结构图谱。
[0035]
图5为pu6grnacas9载体xbai、agei酶切结果胶图。
[0036]
图6为全基因合成的eef1a1-14电泳结果图。
[0037]
图7为pu6grna-eef1a cas9载体的结构图谱。
[0038]
图8为pu6grna-eef1a cas9的agei、bglii酶切胶图结果图。
[0039]
图9为全基因合成的n-nls 1-12电泳结果。
[0040]
图10为pu6grna-eef1a cas9+nnls载体图谱。
[0041]
图11为载体pu6grna-eef1a cas9+nnls的fsei、sbfi酶切胶图。
[0042]
图12为拼接的2727bp片段的胶图结果。
[0043]
图13为改造后载体pu6grna eef1a-mnls-hspcas9-egfp-puro的图谱。
[0044]
图14为dna oligo与pkg-u6grna载体bbsi酶切后粘性末端互补的示意图。
[0045]
图15为pkg-u6grna载体图谱。
[0046]
图16是mstn-grna1插入序列。
[0047]
图17是mstn-grna2插入序列。
[0048]
图18是fndc5-grna1插入序列。
[0049]
图19是fndc5-grna2插入序列。
[0050]
图20为mstn基因编辑效率的对比图。
[0051]
图21为fndc5基因编辑效率的对比图。
具体实施方式
[0052]
本发明所用原始载体px330-u6-chimeric_bb-cbh-hspcas9的结构如图1所示,购自addgene(plasmid#42230,from feng zhang lab)。
[0053]
实施例
[0054]
一、载体改造
[0055]
1、去除grna骨架中多余无效的序列
[0056]
px330-u6-chimeric_bb-cbh-hspcas9(图1)用bbsi、xbai酶切,回收载体片段(约8313bp左右),全基因合成插入片段175bp(seq id no:1),与回收后载体片段重组得到pu6grnacas9载体(图4)。
[0057]
构建具体步骤如下:
[0058]
(1)用限制性内切酶bbsi、xbai酶切px330-u6-chimeric_bb-cbh-hspcas9质粒(酶切体系如表1,37℃反应2h。
[0059]
表1
[0060]
组份量ddh2oto 50ulpx330质粒2ug
10xfd buffer5ulfd bbsi1.5ulfd xbai1.5ul总量50
[0061]
(2)酶切的px330-u6-chimeric_bb-cbh-hspcas9质粒跑琼脂糖胶分离,结果如图2所示,用胶回收试剂盒(诺唯赞fastpure gel dna extraction mini kit#dc301)纯化回收载体大片段,目的片段溶解在50ul ddh2o中,-20℃备用。
[0062]
(3)使用dnaworks设计,全基因合成175bp插入片段,全基因合成引物如表2所示:
[0063]
表2
[0064]
grnasc-1tgtggaaaggacgaaacacc(seq id no:2)grnasc-2tgctatttctagctctaaaacaggtcttctcgaagacccggtgtttcgtcctttccaca(seq id no:3)grnasc-3cctgttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaa(seq id no:4)grnasc-4cacgcgctagaaaaaagcaccgactcggtgccactttttcaagttgataacggactagc(seq id no:5)grnasc-5gtgcttttttctagcgcgtgcgccaattctgcagacaaatggctctagaggtacccgtt(seq id no:6)grnasc-6ttatgtaacgggtacctctagagcc(seq id no:7)
[0065]
全基因合成pcr使用phanta max(诺唯赞p505),按表3所示体系混合:
[0066]
表3
[0067][0068][0069]
pcr条件:95℃3min(95℃15s 58℃15s 72℃20s)循环32次72℃5min;4℃保存。通过全基因合成得到175bp插入片段(seq id no:1),完成pcr后,pcr产物跑1%的琼脂糖电泳,进行分离,结果如图3所示,用胶回收试剂盒(诺唯赞fastpure gel dnaextraction mini kit#dc301)回收目的片段,目的片段溶解在50ul ddh2o中,-20℃备用。
[0070]
(4)使用克隆重组试剂盒(诺唯赞clonexpress ii one step cloning kit#c112)进行载体和175bp插入片段的重组。按表4所示体系加入各组份,混合,37℃反应30min,反应完成立即至于冰上,并用于转化。
[0071]
表4
[0072]
组份体积ul线性化载体150ng插入片段0.04xbp数ng5x ce ii buffer2ulexnase ii1ulddh2oto 10ul
[0073]
(5)转化,克隆送测并质粒小抽
[0074]
1)取100μl dh5α化学感受态细胞(vazyme#c502)置于冰浴中;
[0075]
2)向装有感受态细胞的离心管中加入10μl步骤(4)中得到的重组反应产物,混匀后在冰浴中静置30min;
[0076]
3)将冰浴30min的感受态细胞置于42℃水浴中90s,然后迅速转移到冰浴中,使细胞冷却3min;
[0077]
4)向离心管中加入300μl无菌的lb培养基(不含抗生素),混匀后置于37℃摇床220rpm振荡培养60min;
[0078]
5)将100ul的感受态细胞加到含相应抗生素的lb固体琼脂培养基上,用无菌的涂布棒将感受态细胞涂布均匀;将涂布有感受态细胞的lb固体琼脂培养基倒置于37℃培养箱中培养12~16h。
[0079]
(6)挑取克隆,培养,菌液送测,正确克隆小抽。
[0080]
构建的平板挑取4个克隆,分别置于含300ul的含amp抗性的lb培养基中,37℃培养过夜,第二天分出100ul分别用通用引物lko1_5进行测序,测序结果正确的克隆,分别取菌液20ul到含3ml amp lb的试管中过夜培养,第二天使用质粒小抽试剂盒进行质粒抽提,质粒-20℃保存备用。得到的重组载体pu6grnacas9载体如图4。
[0081]
2、改造启动子及增强子
[0082]
对构建好的pu6grnacas9载体,用xbai和agei内切酶去除启动子(cmv增强子)及增强子序列(chickenβ-actin),回收线性载体序列约7650bp,并合成554bp包含cmv增强子及ef1a启动子的序列(,seq id no:8),与酶切载体pu6grnacas9重组得到pu6grna-eef1a cas9载体(图7)。
[0083]
构建具体步骤如下:
[0084]
(1)用限制性内切酶xbai、agei酶切改造的pu6grnacas9质粒
[0085]
具体实验方法,参看如前所述pu6grnacas9载体的改造过程中载体线性化部分。
[0086]
pu6grnacas9载体xbai、agei酶切胶图结果如图5所示,回收载体大片段。
[0087]
(2)全基因合成554bp插入片段,全基因合成引物如表5所示:
[0088]
表5
[0089][0090][0091]
全基因合成方法参看以上pu6grnacas9载体的改造过程中全基因合成部分。
[0092]
(3)线性化载体和合成插入片段的重组参看以上pu6grnacas9载体的改造过程中
克隆重组部分。eef1a1-14全基因合成电泳结果如图6所示,胶回收目的片段554bp。
[0093]
(4)挑取克隆、培养、菌液送测、正确克隆小抽
[0094]
参看以上pu6grnacas9载体的改造过程中挑取克隆、培养、菌液送测(同样使用通用引物lko1_5进行测序)及正确克隆小抽部分。得到pu6grna-eef1a cas9载体如图7所示。
[0095]
3、cas9基因n端增加nls序列
[0096]
构建好的载体pu6grna-eef1a cas9使用agei、bglii酶切,回收7786bp载体序列,并将增加了nls的序列补充到酶切位点,合成以下序列447bp包括2个核定位信号及部分切除的cas9编码序列(seq id no:23),重组得到pu6grna-eef1a cas9+nnls载体。
[0097]
构建具体步骤如下:
[0098]
(1)用限制性内切酶agei、bglii酶切改造的pu6grna-eef1a cas9质粒
[0099]
具体实验方法,参看以上pu6grnacas9载体的改造过程中载体线性化部分。
[0100]
pu6grna-eef1a cas9的agei、bglii酶切胶图结果如图8所示,回收载体大片段
[0101]
(2)全基因合成447bp插入片段,全基因合成引物如表6所示:
[0102]
表6
[0103]
n-nls-1ccagaacacaggttggaccggtgc(seq id no:24)n-nls-2gatccttgtagtctccgtcgtggtccttatagtccatggtggcaccggtccaacctgtg(seq id no:25)n-nls-3cgacggagactacaaggatcatgatattgattacaaagacgatgacgataagatggccc(seq id no:26)n-nls-4tcttctttggggacccacccacctttcgtttctttttgggggccatcttatcgtcatcg(seq id no:27)n-nls-5ggtgggtccccaaagaagaagcggaaggtcggtatccacggagtcccagcagccgacaa(seq id no:28)n-nls-6cccacagagttggtgccgatgtccaggccgatgctgtacttcttgtcggctgctgggac(seq id no:29)n-nls-7cggcaccaactctgtgggctgggccgtgatcaccgacgagtacaaggtgcccagcaaga(seq id no:30)n-nls-8cttgatgctgtgccggtcggtgttgcccagcaccttgaatttcttgctgggcaccttgt(seq id no:31)n-nls-9gaccggcacagcatcaagaagaacctgatcggagccctgctgttcgacagcggcgaaac(seq id no:32)n-nls-10tatcttcttctggcggttctcttcagccgggtggcctcggctgtttcgccgctgtcgaa(seq id no:33)n-nls-11gagaaccgccagaagaagatacaccagacggaagaaccggatctgctatctgcaagaga(seq id no:34)n-nls-12gccatctcgttgctgaagatctcttgcagatagcagatcc(seq id no:35)
[0104]
全基因合成方法参看以上pu6grnacas9载体的改造过程中全基因合成部分。n-nls 1-12全基因合成电泳结果如图9所示,胶回收447bp目的片段
[0105]
(3)线性化载体和合成插入片段的重组
[0106]
参看以上pu6grnacas9载体的改造过程中克隆重组部分。
[0107]
(4)挑取克隆、培养、菌液送测、正确克隆小抽
[0108]
参看以上pu6grnacas9载体的改造过程中挑取克隆、培养、菌液送测(使用合成引物grna-f:ttttagagctagaaatagcaag进行测序)以及正确克隆小抽部分。得到pu6grna-eef1acas9+nnls载体图谱如图10所示。
[0109]
4、cas9基因c端加入nls、p2a-egfp-t2a-puro、wpre-3’ltr-bgh polya signals
[0110]
以上构建好的载体命名为pu6grna-eef1a cas9+nnls,使用fsei、sbfi酶切,回收载体序列7781bp,合成序列2727bp包括nls-p2a-egfp-t2a-puro-wpre-3’ltr-bgh polya signals(seq id no:36)的序列,与载体片段重组得到载体pu6grna eef1a-mnls-hspcas9-egfp-puro。
[0111]
构建具体步骤如下:
[0112]
(1)用限制性内切酶fsei、sbfi酶切改造的pu6grna-eef1a cas9+nnls质粒,回收
7781bp线性载体片段
[0113]
具体实验方法,参看以上pu6grnacas9载体的改造过程中载体线性化部分。载体pu6grna-eef1a cas9+nnls的fsei、sbfi酶切胶图如图11所示,回收载体大片段。
[0114]
(2)全基因合成2727bp插入片段
[0115]
全基因合成方法参看以上pu6grnacas9载体的改造过程中全基因合成部分。2727bp合成片段来源于3段片段重叠延伸pcr,具体包括
[0116]
片段一:合成含核定位信号编码序列及p2a,egfp重叠序列192bp(seq id no:37),合成引物序列见表7,通过全基因合成得到(方法参看pu6grnacas9载体的改造过程中全基因合成部分)。
[0117]
表7
[0118]
c-nls-1cggcggccacgaaaaaggccggccaggcaaaaaag(seq id no:38)c-nls-2aggccgcttggagccgccctttttcttttttgcctggccggcctttttcgtggccgccg(seq id no:39)c-nls-3ggctccaagcggcctgccgcgacgaagaaagcgggacaggccaagaaaaagaaaggatc(seq id no:40)c-nls-4tccggcttgtttcagcagagagaagtttgttgcgccggatcctttctttttcttggcct(seq id no:41)c-nls-5ctgctgaaacaagccggagatgtcgaagagaatcctggaccggtgagcaagggcgagga(seq id no:42)c-nls-6cggtgaacagctcctcgcccttgctcac(seq id no:43)
[0119]
片段二:egfp片段744bp(seq id no:68),模板为商业化载体egfp-n1,引物为表8,常规pcr得到:
[0120]
表8
[0121]
egfp-fgtgagcaagggcgaggagctgttcaccgg(seq id no:44)egfp-rtagaagacttcccctgccctcgccggagcccttgtacagctcgtccatgccgagagtg(seq id no:45)
[0122]
片段三:t2a-puro-wpre-3’ltr-polya signals序列1840bp(seq id no:69),模板为lenticrisprv2(addgene plasmid#52961),引物如表9,用lenticrisprv2为模板v2-f/v2-r引物pcr,再以前步pcr产物为模板,t2a-f和v2-r pcr得到目的片段1840bp。
[0123]
表9
[0124]
t2a-fgagggcaggggaagtcttctaacatgcggggacgtggaggaaaatcccggccca(seq id no:46)v2-ftgcggggacgtggaggaaaatcccggcccaaccgagtacaagcccacggtgcgcctcg(seq id no:47)v2-rtaccgcatcaggcgcccctgcaggccatagagcccaccgcatccccagcatgcctg(seq id no:48)
[0125]
上述三个片段为模板、使用引物c-nls-1/v2-r pcr得到全长2727bp目的片段(seq id no:36),最终拼接的2727bp片段胶图结果如图12所示。
[0126]
(3)线性化载体和合成插入片段的重组
[0127]
线性化载体pu6grna-eef1a cas9+nnls 7781bp和2727bp插入片段重组,方法参看以上pu6grnacas9载体的改造过程中克隆重组部分。
[0128]
(4)挑取克隆,培养,菌液送测,正确克隆小抽
[0129]
参看以上pu6grnacas9载体的改造过程中挑取克隆,培养,菌液送测(使用合成引物cas9-5-f:ccaccagagcatcaccggcctg(seq id no:49)和f1ori-r:cacacccgccgcgcttaatgcg(seq id no:50)进行测通),正确克隆小抽部分。得到的最终改造载体pu6grna eef1a-mnls-hspcas9-egfp-puro图谱如图14,碱基序列(seq id no:70)。
[0130]
改造后载体pu6grna eef1a-mnls-hspcas9-egfp-puro主要原件:
[0131]
1)grna表达原件:u6-grna scaffold。
u6grna载体图谱如图15所示。
[0156]
pkg-u6grna插入序列(第一段下划线部分为u6启动子序列,大写碱基字母段为两个bbsi酶切位点所在序列,第二段下划线部分为sgrna骨架序列):
[0157]
gataaacatgtgagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgggtcttcgagaagacctgttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttctagcgcgtgcgccaattctgcagacaaatggctctagaggtacccatag(seq id no:67)。
[0158]
每个靶点合成2对互补的dna oligo,通过退火可以形成与pkg-u6grna载体bbsi酶切后粘性末端互补的dna双链(如图14)。
[0159]
合成的mstn基因靶点和fndc5基因靶点的插入序列互补dna oligo如下:
[0160]
mstn-grna1插入序列如图16;mstn-grna2插入序列如图17;fndc5-grna1插入序列如图18;fndc5-grna2插入序列如图19。
[0161]
(1)将grna靶点序列克隆到pkg-u6grna载体骨架上,具体步骤如下:
[0162]
用限制性内切酶bbsi(thermofisher:fd1014)消化1ug pkg-u6grna质粒,37℃反应2h,酶切体系:
[0163][0164]
(2)酶切的pkg-u6grna质粒跑琼脂糖胶分离,用胶回收试剂盒纯化回收酶切产物;
[0165]
(3)公司合成的寡核苷酸链grna-s和grna-a序列按照以下程序退火:
[0166][0167]
95℃,5min然后以5℃/min的速率降至25℃。
[0168]
(4)按照以下体系进行连接反应,37℃反应60min。
[0169][0170]
(5)转化
[0171]
具体方法参看以上pu6grnacas9载体的改造过程中转化,挑取克隆,培养,菌液送测。测序使用通用引物m13f或m13r。
[0172]
正确克隆分别命名为pkg-u6grna(mstn-1),pkg-u6grna(mstn-2),pkg-u6grna(fndc5-1),pkg-u6grna(fndc5-2)质粒小抽,-20℃保存备用。
[0173]
2、电转猪原代细胞
[0174]
分别将cas9表达载体px330或改造的pu6grna eef1a-mnls-hspcas9-egfp-puro与grna表达载体pkg-u6grna(mstn-1)和pkg-u6grna(mstn-2)或pkg-u6grna(fndc5-1)和pkg-u6grna(fndc5-2)共转染猪原代成纤维细胞。
[0175]
使用哺乳动物核转染试剂盒(neon)与neon tm transfection system电转仪进行电转实验。
[0176]
mstn组b:pkg-u6grna(mstn-1)和pkg-u6grna(mstn-2)
[0177]
mstn组330:px330+pkg-u6grna(mstn-1)和pkg-u6grna(mstn-2)
[0178]
mstn组kg:pu6grna eef1a-mnls-hspcas9-egfp-puro+pkg-u6grna(mstn-1)和pkg-u6grna(mstn-2)
[0179]
fndc5组b:pkg-u6grna(fndc5-1)和pkg-u6grna(fndc5-2)
[0180]
fndc5组330:px330+pkg-u6grna(fndc5-1)和pkg-u6grna(fndc5-2)
[0181]
fndc5组kg:pu6grna eef1a-mnls-hspcas9-egfp-puro+pkg-u6grna(fndc5-1)和pkg-u6grna(fndc5-2)
[0182]
1)配制电转反应液,体系如下:
[0183][0184]
混匀过程中注意切勿产生气泡;
[0185]
2)将步骤一制备得到的细胞悬液使用pbs磷酸缓冲液(solarbio)洗一遍,600g离心6min,弃去上清,使用7ul电转基本溶液r重悬细胞,重悬过程中要避免气泡的产生;
[0186]
3)吸取7ul细胞悬液,加入步骤1)中的电转反应液中混匀,混匀过程中注意切勿产生气泡;
[0187]
4)将试剂盒带有的电转杯放置于neon tm transfection system电转仪杯槽内,加入3ml e buffer;
[0188]
5)用电转枪吸取10ul步骤3)得到的混合液,插入点击杯内,选择电转程序(1450v 10ms3pulse),电击转染后立即在超净台内将电转枪中混合液转入到6孔板中,每孔含2ml15%胎牛血清(gibco)+83%dmem培养基(gibco)+1%p/s(gibco penicillin-streptomycin)+1%hepes(solarbio)的完全培养液;
[0189]
6)混匀后放置于37℃,5%co2、5%o2的恒温培养箱中进行培养。
[0190]
7)电转6-12h换液,电转48h使用胰蛋白酶消化并收集细胞到1.5ml ep管中,后期进行突变效率pcr检测。
[0191]
3、pcr检测mstn和fndc5基因突变效率
[0192]
1)向上步收集在1.5ml离心管中的细胞中(根据细胞量,细胞过多需要适当稀释后取部分裂解)加入10ul配置的kapa2g(kapabiosystems:kapa hotstart mouse genotyping kit,货号kk7352))裂解液裂解细胞粗提细胞基因组dna。
[0193]
kapa2g裂解液配制体系如下:
[0194]
10x extract buffer
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1ul
[0195]
kapa express extract enzyme
ꢀꢀꢀꢀ
0.2ul
[0196]
ddh2o
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
8.8ul
[0197]
裂解过程:75℃15min—95℃5min—4℃,反应结束后基因组dna于-20℃保存;
[0198]
2)mstn组使用mstn-f896/mstn-r1351引物进行pcr检测,fndc5组使用fndc5-f209/fndc5-r718引物进行检测,pcr反应体系如下:
[0199][0200]
反应条件如下
[0201][0202]
3)取pcr产物3ul,进行琼脂糖凝胶电泳分析,分析结果如图20和图21所示。图20为mstn基因编辑效率的对比,kg组比330组突变条带(mt,329bp)/野生型条带(wt,456bp)占比更多,说明kg基因编辑效率比330更高。图21为fndc5基因编辑效率的对比,kg组比330组突变条带(mt,421bp)/野生型条带(wt,510bp)占比更多,说明kg基因编辑效率比330更高。
[0203]
根据公式:基因缺失突变效率=100*(mt灰度/mt条带bp数)/(wt灰度/wt条带bp数+mt灰度/mt条带bp数)%,分别计算得到mstn-330组基因缺失突变效率为27.6%,mstn-kg组基因缺失突变效率为86.5%,fndc5-330组基因缺失突变效率为18.6%,fndc5-kg组基因缺失突变效率为81.7%,改造后载体pu6grnaeef1a-mnls-hspcas9-egfp-puro基因编辑效率提高明显(约3~4倍)。
[0204]
综上所述,经过实验验证经过改造的pu6grna eef1a-mnls-hspcas9-egfp-puro载体相对改造前px330载体具有更高的基因编辑效率。其原因是更换了更强的启动子及添加了增强蛋白翻译的元件,提高了cas9的表达,并且增加了核定位信号个数,提高了cas9蛋白的核定位能力。同时,本发明在载体中加入了荧光标记及抗性标记,使其更方便运用于载体阳性转化细胞的筛选及富集。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1