一种富马酸地洛昔醇的制备方法与流程
本发明涉及原料药制备方法技术领域,具体涉及原料药富马酸地洛昔醇的制备方法。
背景技术:
富马酸地洛昔醇(diroximelfumarete,alks-8700,bii098),商品名vumerity,是一种新型口服富马酸药物,用于复发型多发性硬化症(rms)的治疗。富马酸地洛昔醇由alkermes研制,已授权给百健,该药属于免疫抑制剂,每日口服2次,属于控释剂型的富马酸单甲酯(mmf)前药,可在体内快速转化为mmf。
2019年,百健与合作伙伴alkermes联合宣布,双方已向美国食品和药物管理局提交了diroximelfumarate(biib098)的新药申请(nda)。2019年02月25日,fda受理alkermes/百健多发性硬化症药物富马酸地洛昔醇(商品名为vumerity)上市申请,适应症为复发缓解型多发性硬化症(rrms)。
富马酸地洛昔醇英文名为:4-o-[2-(2,5-dioxopyrrolidin-1-yl)ethyl]-1-o-methyl(e)-but-2-enedioate,中文名称:2-(2,5-二氧代吡咯烷-1-基)乙基甲基富马酸酯。cas:1577222-14-0,分子式为c11h13no6,该药物分子中含有(e)-富马酸单甲酯及2,5-二氧代吡咯烷结构单元,其结构式如下:
在2014年,alkermes公司首次报道了富马酸地洛昔醇的合成方法(us8669281b1,wo2014152494a1),并对富马酸地洛昔醇的结构进行专利保护,如下:
其中,r1为未取代的c1-6烷基,l为未取代的c1-6烷基链,未取代的c3-10碳环,未取代的c6-10芳环等。r2或r3为h,c1-6烷基,c2-6烯基等。用反式富马酸单甲酯和n-(2-羟乙基)丁二酰胺经过缩合反应,制备富马酸地洛昔醇,产率为35%。具体路线如下:
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种制备富马酸地洛昔醇的新合成路线,所需物料试剂方便易得、反应产率大大提高、产品易于纯化、杂质易于控制等优点,该制备方法具有明显的技术优势。
为实现上述目的及其他相关目的,本发明的一方面提供一种富马酸地洛昔醇的制备方法,包括如下步骤:
1)氨化反应:将丁二酸酐与氨化试剂进行反应,生成丁二酰亚胺,反应方程式如下:
2)烷基化反应:将丁二酰亚胺与1,2卤代乙烷在催化剂、碱存在的条件下进行反应,生成式iv化合物,反应方程式如下:
式iii化合物中,x选自br、cl、或i,x’选自br、cl、或i;
3)酯化反应:将式iv化合物与反式-富马酸单甲酯进行反应,生成富马酸地洛昔醇,反应方程式如下:
在本发明的一些实施方式中,所述步骤1)中,丁二酸酐与氨化试剂的摩尔比为1:1~1:2。
在本发明的一些实施方式中,其特征在于,所述步骤1)中,氨化试剂选自nh3·h2o、nh2oh·hcl、nh4cl、nh3、nh4oac、尿素中的一种或多种的组合。
在本发明的一些实施方式中,所述步骤1)中还包括如下技术特征的一项或多项:
a1)氨化反应中加入催化剂,所述催化剂为亚磷酸、对甲基苯磺酸、甲磺酸中的一种或多种的组合,所述催化剂的质量为丁二酸酐质量的1~10%;
a2)所述步骤1)中氨化反应的反应温度为120~220℃;
a3)所述步骤1)中,反应后处理的方法为:将反应淬灭、重结晶,即得式ii化合物。
在本发明的一些实施方式中,所述步骤2)中,催化剂为相转移催化剂,所述相转移催化剂选自bu4n+br-、bu4n+i-、苄基三乙基氯化铵、四丁基氯化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵、18冠6中的一种或多种的组合。
在本发明的一些实施方式中,所述步骤2)中,碱选自无机碱,优选选自na2co3、k2co3、koh、naoh中的一种或多种的组合。
在本发明的一些实施方式中,所述步骤2)中,还包括如下技术特征的一项或多项:
b1)所述步骤2)中,反应是在气体保护条件下进行;
b2)所述步骤2)中,丁二酰亚胺与1,2卤代乙烷的摩尔比为1:1~1:3;
b3)所述步骤2)中,所述催化剂的质量为丁二酰亚胺质量的1~10%;
b4)所述步骤2)中,碱与丁二酰亚胺的摩尔比为1~2;
b5)所述步骤2)中,反应是在溶剂存在的条件下进行,所述溶剂选自乙腈,dmf,丙酮,四氢呋喃,1,4-dioxane中的一种或多种的组合;
b6)所述步骤2)中,反应温度为20~90℃。
在本发明的一些实施方式中,所述步骤2)中,反应后处理的方法为:将反应淬灭、萃取、有机相脱溶剂、纯化后即得式iv化合物。
在本发明的一些实施方式中,所述步骤3)中还包括如下技术特征的一项或多项:
c1)所述步骤3)中,式iv化合物与反式-富马酸单甲酯的摩尔比为1:1~1:1.5;
c2)所述步骤3)中,反应是在溶剂条件下进行,所述溶剂包括四氢呋喃、二甲亚砜、1,4-dioxane、水、甲苯、氯仿、丙酮、乙腈中的一种或多种的组合;
c3)所述步骤3)中,反应中使用碱,所述碱为无机碱,优选选自碳酸钠和/或碳酸钾;
c4)所述步骤3)中,碱与式iv化合物的摩尔比为1.0~2.0;
c5)所述步骤3)中,酯化反应的反应温度为20~80℃。
在本发明的一些实施方式中,所述步骤3)中,反应后处理的方法为:将反应淬灭,萃取,脱溶剂、醇溶解,抽滤后即得富马酸地洛昔醇。
附图说明
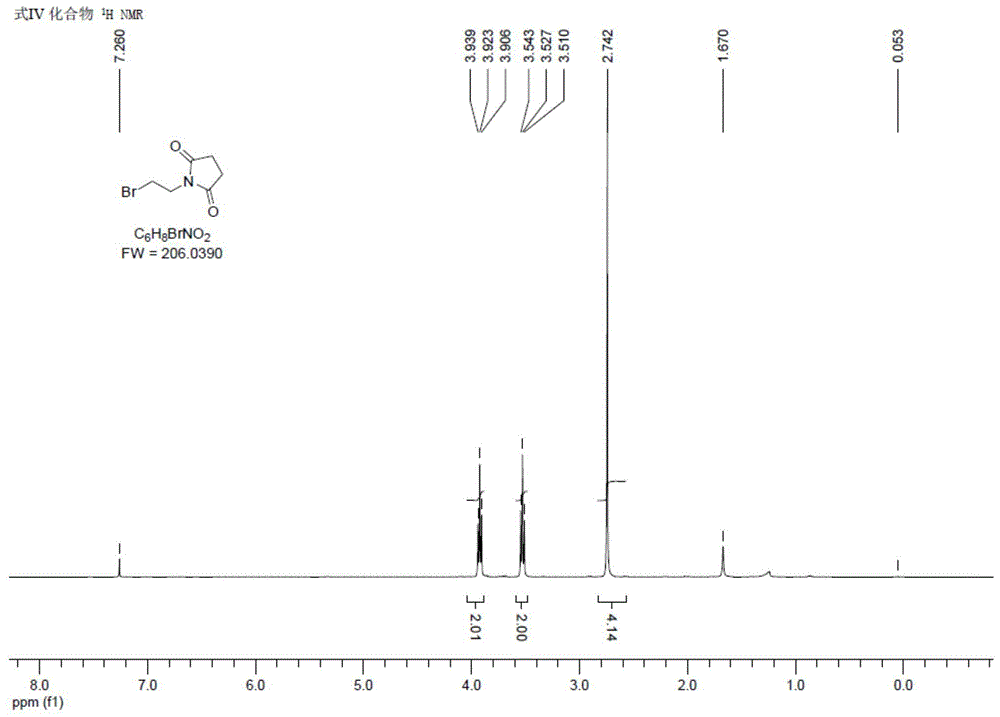
图1显示为本发明实施例1合成得到的式iv化合物的1h-nmr谱图。
图2显示为本发明实施例1合成得到的富马酸地洛昔醇的1h-nmr谱图。
具体实施方式
下面详细说明本发明的富马酸地洛昔醇的制备方法及合成出的富马酸地洛昔醇在治疗复发型多发性硬化症药物中的用途。
本发明第一方面提供一种富马酸地洛昔醇的制备方法,包括如下步骤:
1)氨化反应:将丁二酸酐与氨化试剂进行反应,生成丁二酰亚胺,反应方程式如下:
2)烷基化反应:将丁二酰亚胺与1,2卤代乙烷在催化剂、碱存在的条件下进行反应,生成式iv化合物,反应方程式如下:
式iii化合物中,x选自br、cl、或i,x’选自br、cl、或i;
3)酯化反应:将式iv化合物与反式-富马酸单甲酯进行反应,生成富马酸地洛昔醇,反应方程式如下:
在本发明所提供的制备方法中,所述步骤1)氨化反应中,将丁二酸酐与氨化试剂进行反应,生成丁二酰亚胺,其中丁二酸酐与氨化试剂的摩尔比为1:1~1:2。在一些优选的实施方式中,所述丁二酸酐与氨化试剂的摩尔比为1:1~1:1.2,1:1.2~1:1.4,1:1.4~1:1.6,1:1.6~1:1.8,或1:1.8~1:2。更优选地,所述丁二酸酐与氨化试剂的摩尔比为1:1。所述步骤1)中,氨化试剂选自nh3·h2o、nh2oh·hcl、nh4cl、nh3、nh4oac、尿素中的一种或多种的组合。在一优选实施方式中,所述氨化试剂优选为nh3·h2o。所述步骤1)中,所述氨化反应的温度为120~220℃,这个反应温度是指生成式ii化合物的温度范围。所述氨化反应的温度也可以是120~150℃,150~180℃,180~220℃,180~190℃,190~200℃,200~210℃,或210~220℃。更具体地,在一具体实施方式中,室温下,向所述丁二酸酐中加入氨化试剂,通常情况下,加入氨化试剂会放热,需控制反应温度在60~80℃以内,然后再加入催化剂,然后升温至120~150℃,再经多次升温到180~220℃,得到式ii化合物。所述催化剂选自亚磷酸、对甲基苯磺酸、甲磺酸中的一种或多种的组合,所述催化剂的质量为丁二酸酐质量的1~10%,在本发明的一些具体实施方式中,所述催化剂的质量可以为丁二酸酐质量的1~2%,2~3%,3~4%,4~5%,5~6%,6~7%,7~8%,8~9%,9~10%。本领域技术人员可以根据实际反应情况调整反应时间,例如反应时间为3~12h;在一些具体的实施方式中,氨化反应的反应时间也可以为3~4h,4~5h,5~6h,6~7h,7~8h,8~9h,9~10h,10~11h,或11~12h。反应的后处理的方法包括将反应淬灭、重结晶,即得式ii化合物。
在本发明所提供的制备方法中,所述步骤2)烷基化反应是将丁二酰亚胺与1,2卤代乙烷在催化剂、碱存在的条件下进行反应,生成式iv化合物。式iii化合物中,所述x选自br、cl、i中的一种,所述x’选自br、cl、i中的一种。x和x’的选择可相同,也可不同。所述步骤2)中,所述反应优选是在气体保护条件下进行,所述气体保护可使用的气体包括但不限于氮气、氩气等气体中的一种或多种的组合。
进一步地,所述步骤2)烷基化反应中丁二酰亚胺与1,2卤代乙烷的摩尔比为1:1~1:1.5,在一些优选的实施方式中,所述烷基化应中丁二酰亚胺与1,2卤代乙烷的摩尔比可以为1:1~1:1.2,1:1.2~1:1.3,1:1.3~1:1.4,或1:1.4~1:1.5。
进一步地,所述步骤2)烷基化反应中催化剂为相转移催化剂,所述相转移催化剂选自但不限于bu4n+br-、bu4n+i-、苄基三乙基氯化铵(teba)、四丁基氯化铵、四丁基硫酸氢铵(tbab)、三辛基甲基氯化铵、十二烷基三甲基氯化铵、18冠6中的一种或多种的组合。所述相转移催化剂优选为bu4n+br-。所述催化剂的用量没有特殊限制,只要不对本发明的发明目的产生限制即可,优选的催化剂使用量为:所述催化剂的质量为丁二酰亚胺质量的1~10%,在本发明的一些具体实施方式中,所述催化剂的质量可以为丁二酰亚胺质量的1~2%,2~3%,3~4%,4~5%,5~6%,6~7%,7~8%,8~9%,9~10%。
进一步地,所述烷基化反应中碱选自无机碱,但不限于na2co3、k2co3、koh、naoh中的一种或多种的组合。优选地,所述碱优选为k2co3。所述步骤2)中碱的使用量为:碱与丁二酰亚胺的摩尔比为1~2;在一些优选的实施方式中,所述碱与丁二酰亚胺的摩尔比可以为1~1.2,1.2~1.4,1.4~1.6,1.6~1.8,1.8~2。
进一步地,所述烷基化反应可不使用反应溶剂,也可在反应溶剂中进行,所述反应溶剂优选为有机溶剂,所述有机溶剂包括但不限于乙腈,dmf,丙酮,四氢呋喃,1,4-dioxane中的一种或多种的组合。本领域技术人员可根据反应溶剂的种类、工艺条件和使用环境,调整反应溶剂的用量,具体的使用比例可以为:步骤2)中溶剂的体积与步骤2的反应物的质量的比例为5~10。所述步骤2中的反应物包括n-丁二酰亚胺、1,2-二卤乙烷、催化剂和碱。
进一步地,所述烷基化反应的反应温度为20~90℃。所述烷基化反应的温度也可以是20~30℃,30~40℃,40~50℃,50~60℃,60~70℃,70~80℃,80~90℃。本领域技术人员可以根据实际反应情况调整反应时间,例如反应时间为3~12h;在一些具体的实施方式中,烷基化反应的反应时间也可以为3~4h,4~5h,5~6h,6~7h,7~8h,8~9h,9~10h,10~11h,或11~12h。
本发明的烷基化反应中,反应完成后后处理的具体方法为:将反应淬灭,萃取,有机相脱溶剂、纯化后即得式iv化合物。更具体地,将反应淬灭的方法可以是向反应混合物中加入水淬灭反应,加入水的量例与反应混合物体积比为1:1~1:2。萃取可以选用甲苯萃取多次,例如萃取三次,萃取后合并有机相,洗涤后减压脱除溶剂,可采用饱和食盐水洗涤。纯化步骤可采用将有机相脱溶剂后的残余物使用硅胶柱层析快速纯化得到中间体式iv化合物。产品为无色油状物。
本发明所提供的制备方法中,所述步骤3)的酯化反应中:将式iv化合物与反式-富马酸单甲酯进行反应,生成富马酸地洛昔醇。在酯化反应中,式iv化合物与反式-富马酸单甲酯摩尔比为1:1~1:1.5;所述式iv化合物与反式-富马酸单甲酯的摩尔比可以为1:1~1:1.2,1:1.2~1:1.3,1:1.3~1:1.4,或1:1.4~1:1.5。
进一步地,酯化反应是在溶剂条件下进行,所述溶剂包括但不限于四氢呋喃、二甲亚砜、1,4-dioxane、水、甲苯、氯仿、丙酮、乙腈中的一种或多种的组合;本领域技术人员可根据反应溶剂的种类、工艺条件和使用环境,调整反应溶剂的用量,具体的使用比例可以为:步骤3)中溶剂的体积与步骤3)的反应物的质量的比例为5~10。所述步骤3)中的反应物包括式iv化合物、反式-富马酸单甲酯和碱。
进一步地,酯化反应中使用碱,所述碱选自无机碱,优选为碳酸钠和/或碳酸钾;碱与式iv化合物的摩尔比为1.0~2.0;在一些优选的实施方式中,所述碱与式iv化合物的摩尔比可以为1.0~1.2,1.2~1.4,1.4~1.6,1.6~1.8,1.8~2.0。
进一步地,所述酯化反应的反应温度为20~80℃;所述酯化反应的温度也可以是20~30℃,30~40℃,40~50℃,50~60℃,60~70℃,70~80℃。本领域技术人员可以根据实际反应情况调整反应时间,例如反应时间为3~12h;在一些具体的实施方式中,酯化反应的反应时间也可以为3~4h,4~5h,5~6h,6~7h,7~8h,8~9h,9~10h,10~11h,或11~12h。
本发明的酯化反应中,反应完成后后处理的具体方法为:将反应淬灭,萃取,脱溶剂、醇溶解,抽滤后即得富马酸地洛昔醇。更具体地,将反应淬灭的方法可以是向反应混合物中加入水淬灭反应,加入水的量与反应混合物的体积比为1:1~1:2。萃取可以选用甲苯萃取多次,例如萃取三次,萃取后合并有机相,洗涤后减压脱除溶剂,可采用饱和食盐水洗涤。醇溶解采用甲醇加热溶解,冷却至室温下搅拌过夜。对固体进行抽滤,滤饼用冷甲醇淋洗,固体干燥得到富马酸地洛昔醇。
本发明第二方面提供本发明所述的制备方法制备得到的富马酸洛昔醇。
本发明第三方面提供本发明所述的富马酸洛昔醇在治疗复发型多发性硬化症药物中的用途。
本发明的有益效果:
本发明所需物料试剂方便易得、反应步骤较短总产率比较高、产品通过重结晶易于纯化、基毒杂质通过重结晶易于控制等优点,本发明制备方法具有明显的技术优势。本发明制备方法制得的富马酸地洛昔醇的产率高达81%。
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
在下述实施例中,所使用到的试剂、材料以及仪器如没有特殊的说明,均可商购获得。
实施例1
1、制备丁二酰亚胺(式ii)化合物
向1l的反应瓶中加入丁二酸酐(100g,1.0mol),室温下缓慢滴加nh3-h2o(120ml,1.1mol),控制反应温度在70℃以内。加入催化量亚磷酸(5g,5%),然后升温至120℃下反应1小时,继续升温至190℃下保温6小时。冷却至80℃,加入h2o(100ml),活性炭(3g)脱色。冷却,过滤,乙醇重结晶即得产品为白色固体(84g,收率:85%)。
2、制备中间体式iv化合物
氮气保护下,1l的反应瓶中加入n-丁二酰亚胺(100g,1.0mol)、1,2-二溴乙胺(190g,1.0mol)、bu4nbr(1.0g)、k2co3(140g,1.0mol)和乙腈(600ml)。反应混合物加热至85℃回流至反应12h进行完全。向反应混合物中加入水(300ml)淬灭反应,然后用甲苯萃取三次(500mlx3)。有机相合并,用饱和食盐水洗涤(200ml)洗一次,减压脱除溶剂。残余物使用硅胶柱层析快速纯化得中间体式iv化合物(x=br,182g,收率为88%)为无色油状物。产品直接进行下一步转化。
所得式iv化合物的表征数据:1hnmr(400mhz,dmso-d6):δ=3.92(t,j=6.4hz,2h),3.53(t,j=6.4hz,2h),2.74(s,4h)ppm。
3、制备富马酸地洛昔醇
1000ml三口瓶中,加入中间体式iv化合物(x=br,180g,0.87mol),(e)-富马酸单甲酯(110g,0.87mol),k2co3(130g,0.94mol)以及ch3cn(700ml)。混合物在60℃左右加热保温12小时左右,直至反应进行完全。向反应混合物中加入水(500ml)淬灭反应,然后用甲苯萃取三次(500mlx3)。有机相合并,用饱和食盐水洗涤(300ml)洗一次,减压脱除溶剂。所得固体残留物用甲醇加热溶解,冷却至室温下搅拌过夜。固体抽滤,滤饼用冷甲醇淋洗一次(200ml),所得白色固体置于50℃鼓风干燥得富马酸地洛昔醇(180g,产率:81%)。
所得富马酸地洛昔醇的表征数据:1hnmr(400mhz,dmso-d6):δ=6.82(d,j=4.0hz,2h),4.37(t,j=4.0hz,2h),3.85(t,j=8.0,4.0hz,2h),3.81(s,3h),2.74(s,4h)ppm。
实施例2
1、制备丁二酰亚胺式ii化合物
向1l的反应瓶中加入丁二酸酐(100g,1.0mol),室温下缓慢滴加nh3-h2o(120ml,1.1mol),控制反应温度在60℃以内。加入催化量亚磷酸(5g,5%),然后升温至120℃下反应1小时,继续升温至190℃下保温6小时。冷却至80℃,加入h2o(100ml),活性炭(5g)脱色。冷却,过滤,乙醇重结晶即得产品为白色固体(80g,收率:80%)。
2、制备中间体式iv化合物:
氮气保护下,1l的反应瓶中加入n-丁二酰亚胺(100g,1.0mol)、1,2-二氯乙胺(100g,1.0mol)、bu4ni-(1.0g,1%)、k2co3(140g,1.0mol)和dmf(400ml)。反应混合物加热至85℃至反应12小时。向反应混合物中加入水(500ml)淬灭反应,然后用乙酸乙酯萃取三次(500mlx3)。有机相合并,用饱和食盐水洗涤(300ml)洗一次,减压脱除溶剂。残余物使用硅胶柱层析快速纯化得中间体式iv化合物(x=cl,116g,收率为72%)为无色油状物。产品直接进行下一步转化。
3、制备富马酸地洛昔醇
1000ml三口瓶中,加入中间体式iv化合物(x=br,180g,0.87mol),(e)-富马酸单甲酯(110g,0.87mol),k2co3(130g,0.94mol)以及ch3cn(700ml)。混合物在60℃左右加热保温12小时左右,直至反应进行完全。向反应混合物中加入水(500ml)淬灭反应,然后用乙酸乙酯萃取三次(300mlx3)。有机相合并,用饱和食盐水洗涤(300ml)洗一次,减压脱除溶剂。所得固体残留物用甲醇加热溶解,冷却至室温下搅拌过夜。固体抽滤,滤饼用冷甲醇淋洗一次(150ml),所得白色固体置于50℃鼓风干燥得富马酸地洛昔醇(164g,产率:74%)。
实施例3
1、制备丁二酰亚胺式ii化合物
向1l的反应瓶中加入丁二酸酐(100g,1.0mol),室温下缓慢滴加nh3-h2o(130ml,1.2mol),控制反应温度在60℃以内。加入催化量亚磷酸(5g,5%),然后升温至120℃下反应2小时,继续升温至200℃下保温8小时。冷却至80℃,加入h2o(120ml),活性炭(15g)脱色。冷却,过滤,乙醇重结晶即得产品为白色固体(75g,收率:75%)。
2、制备中间体式iv化合物:
氮气保护下,1l的反应瓶中加入n-丁二酰亚胺(100g,1.0mol)、1-溴-2-二氯乙胺(145g,1.0mol)、bu4n+br-(1.0g,1%)、k2co3(140g,1.0mol)和dioxane(400ml)。反应混合物加热至100℃至反应10小时。向反应混合物中加入水(300ml)淬灭反应,然后用乙酸乙酯萃取三次(300mlx3)。有机相合并,用饱和食盐水洗涤(200ml)洗一次,减压脱除溶剂。残余物使用硅胶柱层析快速纯化得中间体式iv化合物(x=cl,128g,收率为80%)为无色油状物。产品直接进行下一步转化。
3、制备富马酸地洛昔醇
1000ml三口瓶中,加入中间体式iv化合物(x=cl,160g,0.87mol),(e)-富马酸单甲酯(110g,0.87mol),nai(130g,0.87mol),k2co3(130g,0.94mol)以及1,4-dioxane(500ml)。混合物在80℃左右加热保温12小时左右,直至反应进行完全。向反应混合物中加入水(300ml)淬灭反应,然后用甲苯萃取三次(500mlx3)。有机相合并,用饱和食盐水洗涤(300ml)洗一次,减压脱除溶剂。所得固体残留物用甲醇加热溶解,冷却至室温下搅拌过夜。固体抽滤,滤饼用冷甲醇淋洗一次(100ml),所得白色固体置于50℃鼓风干燥得富马酸地洛昔醇(140g,产率:63%)。
实施例4
1、制备丁二酰亚胺式ii化合物
向1l的反应瓶中加入丁二酸酐(100g,1.0mol),室温下缓慢滴加nh3-h2o(110ml,1.0mol),控制反应温度在50℃以内。加入催化量对甲苯磺酸(5g,5%),然后升温至120℃下反应1小时,继续升温至200℃下保温12小时。冷却至60℃,加入h2o(200ml),活性炭(5g)脱色。冷却,过滤,乙醇重结晶即得产品为白色固体(89g,收率:90%)。
2、制备中间体式iv化合物
氮气保护下,1l的反应瓶中加入n-丁二酰亚胺(100g,1.0mol)、1,2-二溴乙胺(190g,1.0mol)、bu4ni(1.0g)、k2co3(140g,1.0mol)和thf(400ml)。反应混合物加热至80℃回流至反应12h进行完全。向反应混合物中加入水(300ml)淬灭反应,然后用甲苯萃取三次(500mlx3)。有机相合并,用饱和食盐水洗涤(100ml)洗一次,减压脱除溶剂。残余物使用硅胶柱层析快速纯化得中间体式iv化合物(x=br,156g,收率为75%)为无色油状物。产品直接进行下一步转化。
3、制备富马酸地洛昔醇
1000ml三口瓶中,加入中间体式iv化合物(x=br,180g,0.87mol),(e)-富马酸单甲酯(110g,0.87mol),nai(130g,0.87mol),k2co3(130g,0.94mol)以及dmf(400ml)。混合物在60℃左右加热保温12小时左右,直至反应进行完全。向反应混合物中加入水(500ml)淬灭反应,然后用甲苯萃取三次(500mlx3)。有机相合并,用饱和食盐水洗涤(300ml)洗一次,减压脱除溶剂。所得固体残留物用甲醇加热溶解,冷却至室温下搅拌过夜。固体抽滤,滤饼用冷甲醇淋洗一次(100ml),所得白色固体置于50℃鼓风干燥得富马酸地洛昔醇(150g,产率:68%)。
上述实施例仅例示性说明本申请的原理及其功效,而非用于限制本申请。任何熟悉此技术的人士皆可在不违背本申请的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本申请所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本申请的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!