一种药用辅料注射级间甲酚的制备方法与流程
本发明属于化合物精制提纯制备领域,特别涉及一种注射级间甲酚的制备方法。
技术背景
三种甲酚异构体中,间甲酚毒性最小,它作为消毒防腐类药物辅料,在一定浓度下可用作肌肉、皮下、皮内注射剂的抗菌防腐剂,在局部用药制剂中也可用作抑菌剂和消毒剂。在精细化工中高纯度的间甲酚一直是化工领域的热门话题,人们不断的在研究如何得到高纯度的间甲酚,然而目前高纯度的间甲酚只是停留在工业级别和原料药物级别,这些间甲酚不能满足药用辅料要求。
工业级别的间甲酚生产:一是以甲苯为原料,经过磺化碱熔合成;另一种是异丙基甲苯法,联产丙酮。在生产维生素e过程中,要求原料药物级别的间甲酚中对甲酚的含量不大于0.2%,间甲酚的含量≥99.8%。上述所产间甲酚在杂质情况、溶剂残留、灼烧残渣等项都存在未知,而药用辅料注射级间甲酚标准要远高于工业级别以及原料药物级别。一个要达到符合注射级别的辅料标准,绝非简单将医药级产品进行灭菌除内毒素即可。尤其是间甲酚这样的有机物辅料,其工艺过程所可能带入的杂质将更为复杂,也更易具有毒性。对于间甲酚在对杂质高度敏感的注射剂中的应用,其纯度的要求甚高(中国药典溶剂要求:正己烷<0.029%,石油醚暂无限度,乙酸乙酯<0.5%,甲苯<0.089%,四氢呋喃<0.072%,n-甲基吡咯烷酮<0.053%)。
目前国内市场上,较多间甲酚药用辅料供应商都是采用从国外原生产厂家采购后对产品分装的方法进行经销。虽然辅料的原生产者可以按照相关标准对产品进行指标的控制,但难以保证经过再次的分装后,产品依然保证原有的品质而不受污染。很多企业分装的过程中并不具备gmp厂房生产的规范条件,分装后不能最大程度保证注射剂用辅料的使用安全性。因此,迫切需要一种药用辅料注射级别的间甲酚的制备方法。
技术实现要素:
为了解决现有国内市场上,药用辅料注射级间甲酚制备方法不规范,间甲酚品质难以保证等问题,本发明提供了一种制备药用辅料注射级间甲酚的方法,采用化学方法进行提纯,通过化学反应将间甲酚结合从而去除杂质,然后析晶、解离晶体,得到注射级间甲酚。该发明能够实现从现有工业级别的间甲酚中提纯,满足药用级辅料标准要求。
为了实现本发明的目的,本发明提供了一种药用辅料注射级间甲酚的制备方法,包括以下步骤:
步骤1:将间甲酚粗品和尿素投入反应釜中,匀速搅拌下升温至80~100,℃保温反应40~90min后,滴加溶剂1,得到反应液;其中,所述尿素与间甲酚的摩尔比为1.20~1.50:1;
步骤2:将步骤1得到的反应液低温析晶1~5h,过滤溶液中析出的固体,将固体用溶剂2反复洗涤不少于2后,真空干燥1~6h,得到络合物固体;
其中,所述的溶剂2选自正己烷、石油醚、丙酮、乙酸乙酯中的一种或两种以上溶剂;
步骤3:将步骤2得到的络合物固体加入纯水中,25~35℃水浴搅拌20~50min,移至室温搅拌1~2h,离心,收集有机层溶液;
步骤4:将步骤3得到的有机层溶液真空干燥后减压精馏,收集≤75℃馏分,得到注射级间甲酚。
其中,步骤1所述的溶剂1选自甲苯、四氢呋喃、n-甲基吡咯烷酮中的任意一种或两种以上溶剂。优选地,溶剂1为甲苯和n-甲基吡咯烷酮的混合液。进一步优选甲苯和n-甲基吡咯烷酮的体积比为1:5~1:1。
在80-100℃下间甲酚充分与尿素络合,而对甲酚和邻甲酚等杂质处于游离状态。在加热保温40-90分钟后滴加溶剂1确保了间甲酚-尿素的络合物与杂质在溶剂中分散开,为后续步骤去除杂质提供较好的条件。甲苯、四氢呋喃、n-甲基吡咯烷酮均能将间甲酚的异构体杂质控制在0.1%以下。
优选地,步骤2所述的低温析晶的温度为-10~-20。℃作为优选方案,步骤2所述的溶剂2为石油醚和乙酸乙酯的混合溶液。进一步优选乙酸乙酯和石油醚的体积比为1:8~1:2。
在低温下析晶1-5h确保了间甲酚-尿素的络合物充分析出,保证了成品的收率。-10~-20℃温度下析晶,成品的收率最高。络合物固体在经过过滤以及溶剂2反复洗涤后将间甲酚异构体对甲酚和邻甲酚充分去除,保证了成品的质量。在洗涤过程中运用低沸点的溶剂正己烷、石油醚、丙酮、乙酸乙酯能有效的去除步骤1中残留的溶剂1,低沸点溶剂经过真空干燥去除,保证了成品质量。
优选地,步骤3所述的络合物固体与纯化水的质量比为1:1~1:5。在此条件下络合物固体水解充分,间甲酚损失量最小。
间甲酚-尿素络合物以氢键作用力存在,水是一种破坏氢键极好的溶剂,络合物经过水解,尿素溶解在水中,间甲酚分离出来,得到粗品间甲酚。间甲酚在水中微溶,经过离心处理,可降低间甲酚损失。
本发明真空干燥去除溶剂以及水分,降低了在减压精馏步骤中难去除溶剂、水分的风险。通过控制馏分温度,提高对残留尿素的控制,保证了成品的质量,达到药用辅料注射级要求。
本发明原料廉价易得、生产成本低,所得产物间甲酚含量≥99.8%、对甲酚控制在≤0.05%、总杂≤0.1%、无溶剂残留、蒸发残渣≤0.05%。其产品的各项指标均满足国内外药典要求(其中,欧洲药典要求:对甲酚和邻甲酚均<0.5%,其它单杂<0.1%,总杂<1.0%,蒸发残渣≤0.1%;美国药典要求:含量98.0%--102.0%,对甲酚和邻甲酚均<0.5%,其它单杂<0.1%,总杂<1.0%;英国药典要求:对甲酚和邻甲酚均<0.5%,其它单杂<0.1%,总杂<1.0%,蒸发残渣≤0.1%;中国药典溶剂要求:正己烷<0.029%,石油醚暂无限度,乙酸乙酯<0.5%,甲苯<0.089%,四氢呋喃<0.072%,n-甲基吡咯烷酮<0.053%),所用溶剂毒性较低,并且可以回收反复使用,实现了废液零排放,减轻了对环境的污染,具有较高的实际应用价值。
附图说明
图1为实施例9注射级间甲酚的气相色谱图;
图2为实施例9注射级间甲酚的气相色谱图的放大图。
具体实施方式
以下结合具体实施例,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
实施例1络合物固体的制备
称取工业级间甲酚(2500g)、尿素(1388.48g)投入到(10l)三口瓶中,置于80-100℃油浴锅中,保温反应1h。反应结束,将反应装置移出油浴锅,加装滴定管,管内装有混合溶剂,其中混合溶剂为:甲苯/n-甲基吡咯烷酮=1/5(甲苯834ml,/n-甲基吡咯烷酮4166ml),搅拌下滴加溶剂。5l溶剂滴毕,取下滴定管,加装带温度计的橡胶塞封闭侧口。
将上述反应瓶移至低温冷却液中,机械匀速搅拌,开启制冷,当冷却液温度降至-15,℃计时结晶约4h。取出反应瓶,过滤,收集固体。用石油醚/乙酸乙酯=5/1(石油醚584ml,乙酸乙酯2917ml)洗涤固体并过滤3次。将收集到的固体置于不锈钢托盘中,放置真空干燥箱中干燥约2h。得到2934g的络合物固体。
实施例2络合物固体的制备
称取工业级间甲酚(2500g)、尿素(2776.96g)投入到(10l)三口瓶中,置于80-100℃油浴锅中,保温反应1h。反应结束,将反应装置移出油浴锅,加装滴定管,管内装有混合溶剂,其中混合溶剂为:甲苯/n-甲基吡咯烷酮=1/1(甲苯3000ml,/n-甲基吡咯烷酮3000ml),搅拌下滴加溶剂。6l溶剂滴毕,取下滴定管,加装带温度计的橡胶塞封闭侧口。
将上述反应瓶移至低温冷却液中,机械匀速搅拌,开启制冷,当冷却液温度降至-15,℃计时结晶约4h。取出反应瓶,过滤,收集固体。用石油醚/乙酸乙酯=2/1(石油醚2333ml,乙酸乙酯1167ml)洗涤固体并过滤4次。将收集到的固体置于不锈钢托盘中,放置真空干燥箱中干燥约2h。得到4176g的络合物固体。
实施例3络合物固体的制备
称取工业级间甲酚(2500g)、尿素(2082.72g)投入到(10l)三口瓶中,置于80-100℃油浴锅中,保温反应1h。反应结束,将反应装置移出油浴锅,加装滴定管,管内装有混合溶剂,其中混合溶剂为:甲苯/n-甲基吡咯烷酮=1/3(甲苯1250ml,/n-甲基吡咯烷酮3750ml),搅拌下滴加溶剂。5l溶剂滴毕,取下滴定管,加装带温度计的橡胶塞封闭侧口。
将上述反应瓶移至低温冷却液中,机械匀速搅拌,开启制冷,当冷却液温度降至-15,℃计时结晶约4h。取出反应瓶,过滤,收集固体。用石油醚/乙酸乙酯=5/1(石油醚2917ml,乙酸乙酯583ml)洗涤固体并过滤3次。将收集到的固体置于不锈钢托盘中,放置真空干燥箱中干燥约2h。得到3483g的络合物固体。
实施例4络合物固体的制备
称取工业级间甲酚(2500g)、尿素(2776.96g)投入到(10l)三口瓶中,置于80-100℃油浴锅中,保温反应1h。反应结束,将反应装置移出油浴锅,加装滴定管,管内装有混合溶剂,其中混合溶剂为:四氢呋喃(3.7l),搅拌下滴加溶剂。3.7l溶剂滴毕,取下滴定管,加装带温度计的橡胶塞封闭侧口。
将上述反应瓶移至低温冷却液中,机械匀速搅拌,开启制冷,当冷却液温度降至-15,℃计时结晶约4h。取出反应瓶,过滤,收集固体。用正己烷(3500ml)洗涤固体并过滤2次。将收集到的固体置于不锈钢托盘中,放置真空干燥箱中干燥约2h。得到4176g的络合物固体。
实施例5络合物固体的制备
称取工业级间甲酚(2500g)、尿素(1388.48g)投入到(10l)三口瓶中,置于80-100℃油浴锅中,保温反应1h。反应结束,将反应装置移出油浴锅,加装滴定管,管内装有混合溶剂,其中混合溶剂为:n-甲基吡咯烷酮7.5l,搅拌下滴加溶剂。7.5l溶剂滴毕,取下滴定管,加装带温度计的橡胶塞封闭侧口。
将上述反应瓶移至低温冷却液中,机械匀速搅拌,开启制冷,当冷却液温度降至-15,℃计时结晶约4h。取出反应瓶,过滤,收集固体。用丙酮(3500ml)洗涤固体并过滤5次。将收集到的固体置于不锈钢托盘中,放置真空干燥箱中干燥约2h。得到2933g的络合物固体。
实施例6络合物固体的制备
称取工业级间甲酚(2500g)、尿素(1944.1g)投入到(10l)三口瓶中,置于80-100℃油浴锅中,保温反应1h。
反应结束,将反应装置移出油浴锅,加装滴定管,管内装有混合溶剂,其中混合溶剂为:甲苯/n-甲基吡咯烷酮=1/1(甲苯2500ml,/n-甲基吡咯烷酮2500ml),搅拌下滴加溶剂。5l溶剂滴毕,取下滴定管,加装带温度计的橡胶塞封闭侧口。
将上述反应瓶移至低温冷却液中,机械匀速搅拌,开启制冷,当冷却液温度降至-15,℃计时结晶约4h。取出反应瓶,过滤,收集固体。用正己烷(3500ml)洗涤固体并过滤3次。将收集到的固体置于不锈钢托盘中,放置真空干燥箱中干燥约2h。得到3895g的络合物固体。
实施例7注射级间甲酚的制备
控制反应釜夹套水浴温度35,℃然后依次向反应釜(20l)中加入纯化水2940ml、实施例1中络合物固体2934g,匀速搅拌40分钟。撤去水浴,反应液室温搅拌1.5小时后停止搅拌,开始离心。离心后下层为有机层(油状物),上层为水层。舍弃水层,合并所有的有机相。有机相加入到2l单口瓶中,加入磁子,在30℃水浴下,搅拌、真空干燥约3h,得到间甲酚有机液。将上步得到的有机液进行减压精馏,控制蒸汽温度在≤75℃收集馏分,馏分在无菌环境下分装,得注射级间甲酚,其气相色谱与图1相似。
实施例8注射级间甲酚的制备
控制反应釜夹套水浴温度35,℃然后依次向反应釜(20l)中加入纯化水25l、实施例2中络合物固体4176g,匀速搅拌40分钟。撤去水浴,反应液室温搅拌1.5小时后停止搅拌,开始离心。离心后下层为有机层(油状物),上层为水层。舍弃水层,合并所有的有机相。有机相加入到2l单口瓶中,加入磁子,在30℃水浴下,搅拌、真空干燥约3h,得到间甲酚有机液。
将上步得到的有机液进行减压精馏,控制蒸汽温度在≤75℃收集馏分,馏分在无菌环境下分装,得注射级间甲酚,其气相色谱与图1相似。
实施例9注射级间甲酚的制备
控制反应釜夹套水浴温度35,℃然后依次向反应釜(20l)中加入纯化水17l、实施例3中络合物固体3483g,匀速搅拌40分钟。撤去水浴,反应液室温搅拌1.5小时后停止搅拌,开始离心。离心后下层为有机层(油状物),上层为水层。舍弃水层,合并所有的有机相。有机相加入到2l单口瓶中,加入磁子,在30℃水浴下,搅拌、真空干燥约3h,得到间甲酚有机液。
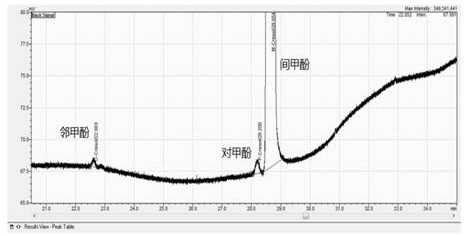
将上步得到的有机液进行减压精馏,控制蒸汽温度在≤75℃收集馏分,馏分在无菌环境下分装,等注射级间甲酚,其气相色谱如图1所示,其放大图如图2所示。
实施例10注射级间甲酚的制备
控制反应釜夹套水浴温度35,℃然后依次向反应釜(20l)中加入纯化水20.8l、实施例4中络合物固体(4176g),匀速搅拌40分钟。撤去水浴,反应液室温搅拌1.5小时后停止搅拌,开始离心。离心后下层为有机层(油状物),上层为水层。舍弃水层,合并所有的有机相。有机相加入到2l单口瓶中,加入磁子,在30℃水浴下,搅拌、真空干燥约3h,得到间甲酚有机液。
将上步得到的有机液进行减压精馏,控制蒸汽温度在≤75℃收集馏分,馏分在无菌环境下分装,得注射级间甲酚,其气相色谱与图1相似。
实施例11注射级间甲酚的制备
控制反应釜夹套水浴温度35,℃然后依次向反应釜(20l)中加入纯化水8.8l、实施例5中络合物固体2933g,匀速搅拌40分钟。撤去水浴,反应液室温搅拌1.5小时后停止搅拌,开始离心。离心后下层为有机层(油状物),上层为水层。舍弃水层,合并所有的有机相。有机相加入到2l单口瓶中,加入磁子,在30℃水浴下,搅拌、真空干燥约3h,得到间甲酚有机液。
将上步得到的有机液进行减压精馏,控制蒸汽温度在≤75℃收集馏分,馏分在无菌环境下分装,得注射级间甲酚,其气相色谱与图1相似。
实施例12注射级间甲酚的制备
控制反应釜夹套水浴温度35,℃然后依次向反应釜(20l)中加入纯化水11.7l、实施例6中络合物固体3895g,匀速搅拌40分钟。撤去水浴,反应液室温搅拌1.5小时后停止搅拌,开始离心。离心后下层为有机层(油状物),上层为水层。舍弃水层,合并所有的有机相。有机相加入到2l单口瓶中,加入磁子,在30℃水浴下,搅拌、真空干燥约3h,得到间甲酚有机液。
将上步得到的有机液进行减压精馏,控制蒸汽温度在≤75℃收集馏分,馏分在无菌环境下分装,得注射级间甲酚,其气相色谱与图1相似。
实施例13杂质的检测
气相色谱法的色谱条件与系统适用性实验:采用直接进样系统程序升温法。色谱柱为30m*250μm,0.5μmcoatingofg7;进样口温度为200℃;氢火焰离子检测器温度为220℃;程序升温:初始温度90℃,保持2min,以30℃/min的速率升至120℃,保持10分钟;最后以3℃/min的速率升至200℃,保持3分钟。以氮气为载气,空气:氢气:氮气=300:30:30,分流比为40:1,流速为每分钟1.8ml。取间甲酚、邻甲酚、对甲酚适量,用甲醇溶解并稀释制成每1ml中各含5mg的溶液,作为系统适应性溶液;精密吸取1μl注入气相色谱仪,要求对甲酚与间甲酚之间的分离度不得小于1.4。
测定方法:分别取实施例7、8、9、10、11、12所得的注射级间甲酚适量,精密称定,用甲醇定量稀释制成每1ml中含10mg的溶液,作为供试品溶液。精密吸取以上稀释剂(甲醇)、供试品溶液各1μl注入气相色谱仪中,记录色谱图。按面积归一化法计算,邻甲酚不得大于0.1%,rrt0.98(对甲酚+杂质f+杂质g)不得大于0.3%,其他单个杂质不得大于0.1%,总杂不得于0.5%。结果见表1。
实施例14含量的检测
气相色谱法的色谱条件与系统适用性实验:采用直接进样系统程序升温法。色谱柱为30m*250μm,0.5μmcoatingofg7;进样口温度为200℃;氢火焰离子检测器温度为220℃;程序升温:初始温度90℃,保持2min,以30℃/min的速率升至150℃,保持0分钟,再以20℃/min的速率升至200℃,保持5分钟。以氮气为载气,空气:氢气:氮气=300:30:30,分流比为40:1,流速为每分钟1.8ml。取苯酚、邻甲酚、对甲酚和间甲酚适量,用甲醇溶解并稀释制成每1ml中含苯酚1mg、邻甲酚5mg、对甲酚5mg及间甲酚5mg的溶液,作为系统适应性溶液;系统适应性溶液图谱中,苯酚、邻甲酚、对甲酚和间甲酚前后分离度均大于1.4。
测定方法:取苯酚适量,加甲醇溶解制成每1ml中含苯酚1mg的溶液,作为内标液;取间甲酚对照品适量,用内标液定量稀释制成每1ml中含1mg的溶液,作为对照品溶液。另分别取实施例7、8、9、10、11、12所得的注射级间甲酚适量,精密称定,用内标液定量稀释制成每1ml中含1mg的溶液,作为供试品溶液。精密量取对照品溶液、供试品溶液各1μl注入气相色谱仪,记录色谱图。按内标法以峰面积计算,即得。结果见表1。
实施例15残留溶剂的检测
分别取实施例7、8、9、10、11、12所得的注射级间甲酚适量,用甲醇定量稀释制成每1ml中含100mg的溶液,作为供试品溶液。另取实施例所用的溶剂1/2适量,精密称定,用甲醇定量稀释制成每1ml中含实施例所用的溶剂1/2适量1mg的溶液,作为对照品溶液。精密吸取以上稀释剂(甲醇)、对照品溶液、供试品溶液各1μl注入气相色谱仪中,照杂质的检测方法测定,记录色谱图。按外标法以峰面积计算,本品含实施例所用的溶剂1/2不得过1%。结果见表1。
实施例16不挥发物的检测
分别取实施例7、8、9、10、11、12所得的注射级间甲酚2.0g,置105℃恒重的蒸发皿中,在水浴上加热使挥发,并在105℃干燥1小时,遗留残渣不得过2mg。结果见表1。
表1
- 还没有人留言评论。精彩留言会获得点赞!