4-((1R,3S)-6-氯-3-苯基-2,3-二氢-1H-茚-1-基)-1,2,2-三甲基哌嗪和4-((1R,3S)-6-氯-3-(苯基-d5)-2,3-二氢-1H-茚-1-基)-2,2-二甲基-1-(甲基-d3)哌嗪的前药的制作方法
4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪和4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的前药
技术领域
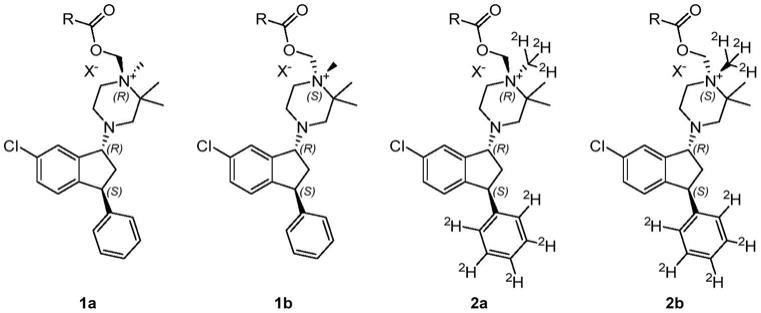
1.本发明涉及:呈1a和1b的形式的4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪,以及呈2a和2b的形式的4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的前药,其中x
‑
是抗衡离子;
[0002][0003]
或其药学上可接受的盐。
[0004]
本发明还提供了包含本发明的前药或其药学上可接受的盐的药物组合物。
背景技术:
[0005]
在wo 93/22293和klaus p.drug hunting[药物搜寻],the medicinal chemistry of 1
‑
piperazino
‑3‑
phenylindans and related compounds[1
‑
哌嗪基
‑3‑
苯基茚满及相关化合物的药物化学],1998,isbn 87
‑
88085
‑
10
‑
4(参见例如第47页表3中和第101页表9a中的化合物69)中已经描述了在哌嗪环的2
‑
和/或3
‑
位被取代的3
‑
芳基
‑1‑
(1
‑
哌嗪基)茚满的一组反式异构体。这些化合物被描述为对于多巴胺d1和d2受体以及5
‑
ht2受体具有高亲和力,并且被建议可用于治疗中枢神经系统的若干种疾病,包括精神分裂症。
[0006]
反式外消旋4
‑
((6
‑
氯
‑3‑
苯基
‑
茚满
‑1‑
基)
‑
1,2,2
‑
三甲基
‑
哌嗪可以例如与在等人,j.med.chem.[药物化学杂志],1995,38,第4380
‑
4392页和wo 93/22293中概括的方法类似地合成。通过拆分反式外消旋4
‑
((6
‑
氯
‑3‑
苯基
‑
茚满
‑1‑
基)
‑
1,2,2
‑
三甲基
‑
哌嗪制造该化合物已经由等人在j.med.chem.[药物化学杂志],1995,38,第4380
‑
4392页中描述,参见表5,化合物(
‑
)
‑
38。所描述的方法包括使用(+)
‑
二甲苯酰基酒石酸用于在乙酸乙酯中拆分,并且将化合物分离为富马酸盐。
[0007]
随后,4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪已经被who给予了国际非专有名称(inn)齐洛那平(zicronapine)。在wo 2005/016900中已经披露了4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪的盐,而后来的专利申请披露了用于制造4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(wo 2011/003423)和拆分同一种化合物(wo 2012/093165)的替代方法。
[0008]4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪是在2a和2b中指示的位置氘(2h)富集的4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪。在wo 2012/176066中已经披露了4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪,其还披露了用于获得4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的合成路线。
[0009]
wo 2014/096151披露了另一种用于经由3,5
‑
二氯
‑1‑
(苯基
‑
d5)茚满的转化获得4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的合成路线。
[0010]
本发明提供了4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪和4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的前药。本发明的前药可以例如改善4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪和4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的摄取、延缓4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪和4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的释放、或降低可能的不良事件的水平。
技术实现要素:
[0011]
本发明提供了化合物,这些化合物是如下所示的前药(1a和1b;2a和2b):
[0012][0013]
及其药学上可接受的盐。
[0014]
1a和2a是在手性氮原子处r
‑
构型的,而1b和2b是在手性氮原子处s
‑
构型的。
[0015]
在一个实施例中,本发明提供了如上所定义的前药或其药学上可接受的盐,其用于在疗法中使用。
[0016]
在一个实施例中,本发明提供了一种药物组合物,其包含如上所定义的本发明的前药或其药学上可接受的盐以及一种或多种药学上可接受的赋形剂。
[0017]
在一个实施例中,本发明提供了如上所定义的前药或其药学上可接受的盐,其用于在用于治疗cns疾病的方法中使用。
[0018]
在一个实施例中,本发明提供了如上所定义的前药或其药学上可接受的盐用于制造用于治疗cns疾病的药剂的用途。
[0019]
在一个实施例中,本发明提供了一种用于治疗cns疾病的方法,所述方法包括向有
需要的患者施用治疗有效量的如上所定义的前药或其药学上可接受的盐。
具体实施方式
[0020]
本发明提供了化合物,这些化合物是如下所示的前药(1a和1b;2a和2b):
[0021][0022]
1a和2a是在手性氮原子处r
‑
构型的,而1b和2b是在手性氮原子处s
‑
构型的。
[0023]
r是直链或支链c1‑
c
11
烷基,诸如甲基;或c3‑
c
10
环烷基,诸如环己基甲基或环己基乙基。
[0024]
附接至4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪的
‑
ch2oc(o)r被称为前药基团。
[0025]
x
‑
选自由以下各项组成的组:卤素阴离子,诸如氯离子、溴离子或碘离子;c1‑
c
10
磺酸根,任选地是氟化的,诸如甲磺酸根、甲苯磺酸根、三氟甲磺酸根或九氟丁烷磺酸根;以及直链或支链c1‑
c
11
羧酸根,任选地是氟化的,诸如三氟乙酸根。
[0026]
化合物1a和1b具有天然氢同位素分布,而2a和2b在指定位置富集氘(2h)。
[0027]
前药
[0028]
前药通常是本身可以不具有任何药理学活性、但在向患者施用时被代谢以提供药理学活性化合物的化合物。更具体地,本发明的前药是在向患者施用时被代谢以提供4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(前药1a和1b)或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(前药2a和2b)的化合物。
[0029]
盐
[0030]
可以将本发明的一些前药作为药学上可接受的酸加成盐提供。术语药学上可接受的盐包括与无机酸和/或有机酸(诸如盐酸、氢溴酸、磷酸、亚硝酸、硫酸、苯甲酸、柠檬酸、葡萄糖酸、乳酸、马来酸、琥珀酸、酒石酸、乙酸、丙酸、草酸、马来酸、富马酸、谷氨酸、焦谷氨酸、水杨酸、水杨酸、糖精和磺酸(诸如甲磺酸、乙磺酸、甲苯磺酸和苯磺酸))形成的盐。以上列出的一些酸是二元酸或三元酸,即含有两个或三个酸性氢的酸(诸如磷酸、硫酸、富马酸和马来酸)。二元酸和三元酸可以形成1:1、1:2或1:3(三元酸)盐,即两个或三个本发明化合物分子与一个酸分子之间形成的盐。
[0031]
形成药学上可接受的盐的有用的酸和碱的另外实例可以例如在stahl和wermuth(编辑)“handbook of pharmaceutical salts.properties,selection,and use[药用盐手
册:特性、选择和使用]”,威利
‑
vch出版社(wiley
‑
vch),2008中找到。
[0032]
治疗有效量
[0033]
在本发明上下文中,术语化合物的“治疗有效量”意指足以在包括施用所述化合物的治疗性介入中治愈、缓解或部分阻滞给定疾病及其并发症的临床表现的量。将足以将其实现的量定义为“治疗有效量”。用于各目的有效量将取决于疾病或损伤的严重程度以及受试者的体重及一般状态。典型地施用本发明的前药以实现与施用1
‑
60mg 4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(诸如1
‑
30mg 4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪,诸如5mg、10mg、15mg或20mg(以游离碱计算))可比较的治疗效果。这意指:例如“20mg 4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪”意指20mg 4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪游离碱,其中实际施用量必须针对前药基团的重量进行调节并且进一步针对抗衡离子的重量进行调节。
[0034]
在本发明上下文中,“治疗(treatment)”或“治疗(treating)”旨在指示为了缓解、阻滞、部分阻滞或延迟疾病的临床表现的进展或者治愈疾病的目的而管理和护理患者。待治疗的患者优选是哺乳动物,特别是人类。
[0035]
疾病
[0036]
在本发明上下文中,“cns疾病”旨在指示中枢神经系统的疾病。
[0037]
如例如wo 2005/016900和wo 2012/176066中所披露的,预期4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪和4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(即,1a、1b、2a和2b的母体形式)的药理学特性也使得化合物可用于治疗cns疾病,这些cns疾病包括但不限于精神病、特别是精神分裂症或涉及精神病症状的其他疾病,诸如例如精神分裂症、难治性精神分裂症(trs)、精神分裂症样障碍、分裂情感性障碍、妄想性障碍、短暂性精神病性障碍、共有型精神病性障碍以及存在精神病性症状的其他精神病性障碍或疾病,例如双相性障碍,诸如双相性障碍中的躁狂症。本发明的化合物和/或组合物可以进一步使用于治疗障碍,诸如描述在例如以下项中的那些:美国专利号5,807,855;7,648,991;7,767,683;7,772,240;8,076,342;美国专利公开号2008/0269248;2010/0069676;2011/0178094;2011/0207744;wo 2005/016900;ep 0 638 073;以及j.med.chem.[药物化学杂志]1995,38,4380
‑
4392;将每个通过引用以其整体并入本文。本发明还涉及本发明的化合物作为与其他治疗剂(诸如描述在例如以下项中的那些)结合的组合疗法的医学用途:美国专利号5,807,855;7,648,991;7,767,683;7,772,240;8,076,342;美国专利公开号2008/0269248;2010/0069676;2011/0178094;2011/0207744;wo 2005/016900;ep 0 638 073;以及j.med.chem.[药物化学杂志]1995,38,4380
‑
4392;将每个通过引用以其整体并入本文。
[0038]
在本发明上下文中,难治性精神分裂症旨在表明尽管用足够剂量和持续时间的抗精神病药进行了两次治疗,但仍缺乏令人满意的临床改善。
[0039]
药物组合物
[0040]
药物组合物可以被具体配制以通过任何合适的途径施用,该合适的途径诸如口服、经直肠、经鼻、经颊、舌下、经皮和肠胃外(例如皮下、肌内和静脉内)途径;口服途径是优选的。
[0041]
应当理解,该途径将取决于待治疗的受试者的一般状况和年龄、待治疗的病症的性质以及活性成分。
[0042]
在本发明上下文中,术语“赋形剂”或“药学上可接受的赋形剂”是指药用赋形剂,其包括但不限于填充剂、抗粘合剂、粘合剂、包衣、着色剂、崩解剂、调味剂、助流剂、润滑剂、防腐剂、吸着剂、甜味剂、溶剂、媒介物和佐剂。
[0043]
本发明还提供了一种药物组合物,其包含4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪前药(1a和1b)或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪前药(2a和2b)或其药学上可接受的盐。根据本发明的药物组合物可以用药学上可接受的赋形剂根据常规技术进行配制,这些常规技术诸如在remington,the science and practice of pharmacy[雷明顿,药学科学与实践],第22版(2012),allen,loyd v.,jr.编中披露的那些。
[0044]
用于口服施用的药物组合物包括固体口服剂型,诸如片剂、胶囊、粉剂以及颗粒剂;和液体口服剂型,诸如溶液、乳液、悬浮液和糖浆以及待溶解或悬浮在适当液体中的粉剂和颗粒剂。
[0045]
固体口服剂型可以呈现为离散单位(例如,片剂或硬胶囊或者软胶囊),这些离散单位各自含有预定量的活性成分以及优选地一种或多种合适的赋形剂。适当时,根据本领域中熟知的方法,固体剂型可以被制备为具有包衣,诸如肠溶衣,或者它们可以被配制以提供活性成分的修饰释放,诸如延迟释放或延长释放。适当时,该固体剂型可以是在唾液中崩解的剂型,例如像口腔分散片剂。
[0046]
适用于固体口服制剂的赋形剂的实例包括但不限于:微晶纤维素、玉米淀粉、乳糖、甘露醇、聚维酮、交联羧甲基纤维素钠、蔗糖、环糊精、滑石、明胶、果胶、硬脂酸镁、硬脂酸和纤维素的低级烷基醚。类似地,固体制剂可以包含本领域已知的用于延迟或延长释放制剂的赋形剂,诸如单硬脂酸甘油酯或羟丙基甲基纤维素。
[0047]
如果将固体材料用于口服施用,则该制剂可以例如通过将活性成分与固体赋形剂混合并且随后在常规压片机中压缩混合物来制备;或可以例如将该制剂以例如粉剂、丸剂或微型片剂形式置于硬胶囊中。固体赋形剂的量将广泛变化,但将典型地在从约25mg至约1g/剂量单位的范围内。
[0048]
液体口服剂型可以呈现为例如酏剂、糖浆、口服滴剂或充液胶囊。液体口服剂型还可以呈现为粉剂,用于在水性或非水性液体中的溶液或悬浮液。适用于液体口服制剂的赋形剂的实例包括但不限于乙醇、丙二醇、甘油、聚乙二醇、泊洛沙姆、山梨醇、聚山梨醇酯、甘油单酯和甘油二酯、环糊精、椰子油、棕榈油和水。液体口服剂型可以例如通过将活性成分溶解或悬浮在水性或非水性液体中或通过将活性成分掺入水包油或油包水液体乳液中来制备。
[0049]
可以将另外的赋形剂(诸如,着色剂、调味剂和防腐剂等)用于固体和液体口服制剂中。
[0050]
用于肠胃外施用的药物组合物包括:用于注射或输注的无菌水性及非水性溶液、分散液、悬浮液或乳液,用于注射或输注的浓缩物以及待在使用之前在用于注射或输注的无菌溶液或分散液中重构的无菌粉剂。适用于肠胃外制剂的赋形剂的实例包括但不限于水、椰子油、棕榈油和环糊精溶液。必要时应该适当缓冲水性制剂,并且用足够的盐水或葡萄糖使其等张。
[0051]
其他类型的药物组合物包括栓剂、吸入剂、乳膏剂、凝胶剂、皮肤贴剂、植入物和用于经颊或舌下施用的制剂。
[0052]
必要条件是用于任何药物制剂的赋形剂符合预期的施用途径并且与活性成分相容。
[0053]
本发明的实施例
[0054]
在以下实施例中进一步描述本发明:
[0055]
1.一种4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(呈1a和1b的形式)或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(呈1a和1b的形式)的前药
[0056][0057]
其中x
‑
是选自由以下各项组成的组的抗衡离子:卤素阴离子,诸如氯离子、溴离子或碘离子;c1‑
c
10
磺酸根,任选地是氟化的,诸如甲磺酸根、甲苯磺酸根、三氟甲磺酸根或九氟丁烷磺酸根;以及直链或支链c1‑
c
11
羧酸根,任选地是氟化的,诸如三氟乙酸根;或其药学上可接受的盐。
[0058]
2.根据实施例1所述的前药,其中r选自由以下各项组成的组:直链或支链c1‑
c
11
烷基和c3‑
c
10
环烷基,或其药学上可接受的盐。
[0059]
3.根据实施例1和2中任一项所述的前药,其中r选自由以下各项组成的组:甲基、叔丁基、正十一烷和环己基甲基,或其药学上可接受的盐。
[0060]
4.根据实施例1至3中任一项所述的前药,其中所述药学上可接受的盐由以下形成:盐酸、氢溴酸、磷酸、亚硝酸、硫酸、苯甲酸、柠檬酸、葡萄糖酸、乳酸、马来酸、琥珀酸、酒石酸、乙酸、丙酸、草酸、马来酸、富马酸、谷氨酸、焦谷氨酸、水杨酸、水杨酸、糖精和磺酸诸如甲磺酸、乙磺酸、甲苯磺酸和苯磺酸。
[0061]
5.根据实施例1至4中任一项所述的前药,其选自由以下各项组成的组:(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓、(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓、(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓、(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓、(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓、(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓、(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓、(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓、(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓、(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓、(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓、(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓、(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓、(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓、(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓、(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓,其中的每一个与选自由以下各项组成的组的抗衡离子组合:卤素阴离子,诸如氯离子、溴离子或碘离子;c1‑
c
10
磺酸根,任选地是氟化的,诸如甲磺酸根、甲苯磺酸根、三氟甲磺酸根或九氟丁烷磺酸根;以及直链或支链c1‑
c
11
羧酸根,任选地是氟化的,诸如三氟乙酸根;或其药学上可接受的盐。
[0062]
6.一种药物组合物,其包含根据实施例1至5中任一项所述的前药或其药学上可接受的盐以及一种或多种药学上可接受的赋形剂。
[0063]
7.根据实施例1至5中任一项所述的化合物或其药学上可接受的盐或者根据实施例6所述的药物组合物,其用于在疗法中使用。
[0064]
8.根据实施例1至5中任一项所述的化合物或其盐或者根据实施例6所述的药物组合物用于制造用于治疗中枢神经系统(cns)疾病的药剂的用途。
[0065]
9.根据实施例1至5中任一项所述的化合物或者根据实施例6所述的药物组合物,其用于在用于治疗cns疾病的方法中使用。
[0066]
10.一种用于治疗cns疾病的方法,所述方法包括向有需要的患者施用治疗有效量的根据实施例1至5中任一项所述的化合物或者根据实施例6所述的药物组合物。
[0067]
实例
[0068]
本发明的化合物
[0069]
本发明的化合物可以由4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(1)或4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(2)制备,这两者均描述在如上所讨论的现有技术中:
[0070][0071]
可以通过用烷基化剂3的处理直接地或经由用更大反应性的卤素进行y的原位芬克尔斯坦(finkelstein)置换(诸如通过添加碘化钠从y=cl转化为y=i或者通过添加四正丁基溴化铵或溴化钠从y=cl转化为y=br)来将这些化合物在非苄基哌嗪氮原子上烷基化。取决于是否使用1或2作为底物,反应将提供n
‑
非对映异构体1a/1b或2a/2b的混合物。可以通过重结晶和/或通过手性色谱法(使用高效液相色谱法(hplc)、超临界流体色谱法(sfc)、或模拟移动床色谱法(smb))分离这些n
‑
非对映异构体。可以使用例如离子交换树脂将阴离子y
‑
与x
‑
交换。y
‑
可以选自与如上所定义的x
‑
相同的列表。最后两个操作的顺序可以颠倒,并且两个步骤都可以执行若干次。烷基化剂3是可商购的,诸如来自combiblocks的3b(目录号qc
‑
7757),或者可以以如说明书中针对3a所描述的或如文献中所描述的(对于磺酸盐,参见例如wo 2012/137225)类似的方式制备。
[0072]
用于制备本发明的前药的方法。
[0073]
对于高效液相色谱法(hplc)、液相色谱/质谱法(lc/ms)、超临界流体色谱法(sfc)、以及离子交换法,使用以下方法。
[0074]
方法1:在配备有agela innoval ods 250
×
80mm柱(10微米粒度;柱温20℃)的agela
‑
hp
‑
q
‑
p600仪器上执行hplc。洗脱液:0.05%hcl水溶液/乙腈4:1至1:1,在20min内,以150ml/min的流速。
[0075]
方法2:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&1956a仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中的0.018%三氟乙酸1:0,持续0.8min;1:0至2:3,在6.2min内;2:3,持续3min;2:3至1:0,在0.1min内,以0.6ml/min的流速。
[0076]
方法3:在配备有phenomenex luna c18 250
×
50mm柱(10微米粒度;柱温20℃)的agela
‑
hp
‑
q
‑
p600仪器上执行hplc。洗脱液:0.05%hcl水溶液/乙腈4:1至1:1,在20min内,以80ml/min的流速。
[0077]
方法4:在配备有在40℃下操作的chiralpak ad
‑
h 250
×
30mm柱(粒度5微米)的thar sfc80制备型仪器上以堆叠注入(stacked injection)执行sfc。洗脱液:co2/在甲醇
中的0.1%三氟乙酸7:3,持续5min,以70g/min的流速。系统背压100巴。
[0078]
方法5:在配备有chiralpak ad
‑
3 100
×
4.6mm柱(粒度3微米;柱温40℃)的thar analytical sfc仪器上执行sfc。洗脱液:co2/在甲醇中的0.05%异丙胺95:5至3:2,在5min内,以4.0ml/min的流速。系统背压100巴。
[0079]
方法6:如方法4执行sfc,运行时间为7min。
[0080]
方法7:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&ms 1956a仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中的0.018%三氟乙酸9:1,持续0.4min;9:1至0:1,在3.0min内;0:1,持续0.45min;0:1至9:1,在0.01min内;9:1,持续0.64min,以0.8ml/min的流速。
[0081]
方法8:如方法3执行hplc,但以在20min内3:2至3:7的洗脱液梯度。
[0082]
方法9:在配备有chiralpak ad
‑
h 250
×
30mm柱(粒度5微米;柱温40℃)的thar sfc80制备型仪器上以堆叠注入执行sfc。洗脱液:co2/在甲醇中的0.1%三氟乙酸4:1,持续6min,以70g/min的流速。系统背压100巴。
[0083]
方法10:在配备有chiralpak ad
‑
3 150
×
4.6mm柱(粒度3微米;柱温35℃)的waters acquity upc2仪器上执行sfc。洗脱液:co2/在甲醇中的0.05%异丙胺3:2,在6min或10min内,以2.5ml/min的流速。系统背压1500psi。
[0084]
方法11:使用呈氯化物形式(717;内部(domestic)10024160)离子交换树脂的阴离子交换树脂执行离子交换。将样品溶解在乙腈和水的1:1混合物中,并且允许溶液缓慢通过具有树脂的柱(使用约六倍于样品的量的树脂)。添加乙腈/水(1:1)直至所有样品都被洗脱。将合并的产物级分在真空中浓缩并且冻干以提供一种或多种化合物的氯化物盐。
[0085]
方法12:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&ms 1956a仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中的0.018%三氟乙酸99:1至1:9,在3.4min内;1:9至0:1,在0.45min内;0:1至99:1,在0.01min内;991:1,持续0.64min,以0.8ml/min的流速。
[0086]
方法13:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&ms 1956a仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中的0.018%三氟乙酸3:2至0:1,在3.4min内;0:1,持续0.45min;0:1至3:2,在0.01min内;3:2,持续0.64min,以0.8ml/min的流速。
[0087]
方法14:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&ms 6120仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中的0.018%三氟乙酸3:1至0:1,在3.4min内;0:1,持续0.45min;0:1至3:1,在0.01min内;3:1,持续0.64min,以0.8ml/min的流速。
[0088]
方法15:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&ms 6120b仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中的0.018%三氟乙酸9:1至1:4,在4.0min内;1:4,持续2.0min;1:4至9:1,在0.01min内;9:1,持续2.0min,以0.8ml/min的流速。
[0089]
方法16:如方法9执行sfc,运行时间为8min。
[0090]
方法17:在配备有phenomenex luna c18(2)50
×
2mm柱(5微米粒度;柱温40℃)的agilent 1200&ms 1956a仪器上执行lc/ms。洗脱液:在水中的0.037%三氟乙酸/在乙腈中
的0.018%三氟乙酸1:0,持续0.8min;1:0至2:3,在6.2min内;2:3,持续3min;2:3至1:0,在0.1min内,以0.8ml/min的流速。
[0091]
方法18:在配备有在25℃下的agela 40
×
10cm(20
‑
40nm)c18柱的biotage one仪器上执行反相柱色谱法。洗脱液:水和0.4%浓盐酸7:3至3:7,在30min内;3:7,持续25min;0:1,持续10min,以120ml/min的流速。
[0092]
实例1:乙酸碘甲酯的制备(烷基化剂3a)
[0093][0094]
在黑暗中在20℃下向nai(29.0g)和乙腈(140ml)的混合物中逐滴添加乙酸氯甲酯(3a1;20.0g)。将反应在环境温度下搅拌24小时。将所得混合物在甲基叔丁基醚(mtbe;160ml)与水(200ml)之间分配,并且将水层用mtbe(150ml)萃取。将合并的有机层依次用饱和碳酸氢钠水溶液(200ml)、10%亚硫酸钠水溶液(200ml)、以及饱和氯化钠水溶液(100ml)洗涤,然后将其经硫酸钠干燥、过滤并且浓缩以提供乙酸碘甲酯(3a;15.5g),其对于下一步骤是足够纯的。
[0095]
实例2:正十二烷酸碘甲酯的制备(烷基化剂3c)
[0096][0097]
在氩气气氛下在
‑
10℃下将正十二烷酰氯(3c2;80.0g)缓慢添加至多聚甲醛(22.0g)和氯化锌(24.9g)在乙腈(550ml)中的混合物中。将反应混合物在该温度下搅拌1小时并且在环境温度下搅拌18小时。将粗反应混合物通过柱快速色谱法在硅胶上直接纯化(梯度洗脱液:石油醚/乙酸乙酯50/1至2/1),以提供正十二烷酸氯甲酯(3c1;45.4g),其对于下一步骤是足够纯的。在黑暗中在环境温度下向碘化钠(32.5g)在乙腈(300ml)中的混合物中逐滴添加化合物3c1(45.0g)。将反应在环境温度下搅拌24小时。将粗混合物分配在mtbe(300ml)与水(300ml)之间,并且将水层用mtbe(100ml)萃取。将合并的有机层依次用饱和碳酸氢钠水溶液(200ml)、10%亚硫酸钠水溶液(150ml)、以及饱和氯化钠水溶液(150ml)洗涤,然后将其经硫酸钠干燥、过滤并且浓缩以提供正十二烷酸碘甲酯(3c;51.0g),其对于下一步骤是足够纯的。
[0098]
实例3:2
‑
环
‑
己基乙酸碘甲酯的制备(烷基化剂3d)
[0099][0100]
在氩气气氛下将亚硫酰氯(69ml)添加至2
‑
环
‑
己基乙酸(3d3;44.8g)在甲苯(180ml)中的溶液中,并且将混合物在110℃下搅拌12小时。在真空中去除挥发物以提供2
‑
环
‑
己基乙酰氯(3d2;42.3g),其对于下一步骤是足够纯的。在氩气气氛下在
‑
10℃下将2
‑
环
‑
己基乙酰氯(3d2;48.6g
‑
来自前一步骤和另一批次的合并材料)添加至氯化锌(20.6g)和多聚甲醛(18.2g)在乙腈(336ml)中的溶液中。将反应混合物在
‑
10℃下搅拌1小时,并且
然后在环境温度下搅拌18小时。在真空中去除挥发物。将残留物通过柱快速色谱法在硅胶上直接纯化(梯度洗脱液:石油醚/乙酸乙酯1/0至0/1),以提供2
‑
环
‑
己基乙酸氯甲酯(3d1;24.0g),其对于下一步骤是足够纯的。在黑暗中在环境温度下向碘化钠(18.1g)在乙腈(126ml)中的溶液中逐滴添加2
‑
环
‑
己基乙酸(3d1;21.9g)。将反应在环境温度下搅拌24小时。将粗混合物分配在mtbe(200ml)与水(200ml)之间,并且将水层用mtbe(150ml)萃取。将合并的有机层依次用饱和碳酸氢钠水溶液(200ml)、10%亚硫酸钠水溶液(200ml)、以及饱和氯化钠水溶液(100ml)洗涤,然后将其经硫酸钠干燥、过滤并且浓缩以提供2
‑
环
‑
己基乙酸碘甲酯(3d;27.0g),其对于下一步骤是足够纯的。
[0101]
实例4:乙酰氧基甲基1a和1b的制备
[0102][0103]
将4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(1;6.0g)在乙腈(42ml)中的溶液加热至80℃,然后添加乙酸碘甲酯(3a;6.76g)。将混合物在80℃下搅拌1小时,然后在真空中去除挥发物。将残留固体悬浮于mtbe(25ml)中,并且然后滤出。将滤饼用mtbe(15ml)洗涤并且干燥以提供约8g的(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓碘化物(1a1;x=i
‑
)和(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓碘化物(1b1;x=i
‑
)的混合物。通过离子交换(方法11)将该材料从碘化物盐转化为相应的氯化物盐以提供以下的粗氯化物盐:1a1;x=cl
‑
和1b1;x=cl
‑
。通过制备型hplc(方法1)纯化该材料以给出(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物和(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物的混合物(1a1;x=cl
‑
和1b1;x=cl
‑
;3g)。
[0104]1h nmr:400mhz甲醇
‑
d
4 7.56(宽峰s,1h),7.29(t,j=7.2hz,3h),7.25
‑
7.18(m,1h),7.14
‑
7.09(m,2h),6.96(d,j=7.9hz,1h),5.50(宽峰s,1h),5.44
‑
5.33(m,1h),4.72(宽峰s,1h),4.60
‑
4.48(m,1h),3.78(宽峰d,j=5.7hz,1h),3.66(宽峰s,1h),3.19
‑
2.81(m,8h),2.26(s,4h),1.71
‑
1.57(m,6h)。
[0105]
lc/ms(方法17):保留时间6.60min,86.5%uv纯度(220nm),m/z质量观测值427.1。
[0106]
实例5:乙酰氧基甲基2a和2b的制备
[0107][0108]
以类似于1a1和1b1的方式制备(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓碘化物和(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓碘化物(2a1;x=i
‑
和2b1;x=i
‑
)以及(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物和(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a1;x=cl
‑
和2b1;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(2;6.50g)和在乙腈(45.0ml)中的乙酸碘甲酯(3a;7.16g),以提供约8g粗碘化物和2.00g(r)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物和(s)
‑1‑
(乙酰氧基甲基)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a1;x=cl
‑
和2b1;x=cl
‑
)的hplc纯化(方法1)混合物。
[0109]1h nmr:400mhz甲醇
‑
d
4 7.61
‑
7.54(m,1h),7.34
‑
7.28(m,1h),6.96(d,j=8.3hz,1h),5.51(宽峰d,j=12.7hz,1h),5.47
‑
5.35(m,1h),4.74(宽峰s,1h),4.56(宽峰s,1h),3.77(宽峰s,1h),3.68(宽峰s,1h),3.27
‑
2.78(m,5h),2.26(d,j=0.9hz,4h),1.73
‑
1.58(m,6h)。
[0110]
lc/ms(方法17):保留时间6.56min,93.5%uv纯度(220nm),m/z质量观测值435.2。
[0111]
实例6:新戊酰氧基甲基1a和1b的制备
[0112][0113]
以类似于1a1和1b1的方式制备(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓碘化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓碘化物(1a2;x=i
‑
和1b2;x=i
‑
)以及(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(1a2;x=cl
‑
和1b2;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(1;4.6g)和在乙腈(32ml)中的新戊酸碘甲酯(3b;6.27g),以提供约5g粗碘化物和约4g(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(1a2;x=cl
‑
和1b2;x=cl
‑
)的hplc纯化(方法3)混合物。将该混合物通过手性sfc(方法4)分离。如之前使用方法11和3纯化两种产物以提供两种产物:
[0114]
第一洗脱异构体:1.00g的(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(1a2;x=cl
‑
)或(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(1b2;x=cl
‑
)。
[0115]1h nmr:400mhz甲醇
‑
d
4 7.71(宽峰s.,1h),7.39
‑
7.27(m,3h),7.26
‑
7.19(m,1h),7.18
‑
7.10(m,2h),6.97(d,j=8.4hz,1h),5.70
‑
5.57(m,1h),5.52(d,j=8.8hz,1h),4.93(宽峰s.,1h),4.69(t,j=7.6hz,1h),3.85(宽峰s.,2h),3.50
‑
3.31(m,4h),3.16(s,3h),3.05
‑
2.92(m,1h),2.42
‑
2.28(m,1h),1.71(宽峰s,6h),1.31(s,9h)。
[0116]
lc/ms(方法12):保留时间3.12min,98.5%uv纯度(220nm),m/z质量观测值469.2。
[0117]
sfc(方法5):保留时间1.91min,>99%uv纯度(220nm)。
[0118]
第二洗脱异构体:0.80g的(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(1b2;x=cl
‑
)或(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(1a2;x=cl
‑
)。
[0119]1h nmr:400mhz甲醇
‑
d
4 7.69(宽峰s.,1h),7.37
‑
7.26(m,3h),7.26
‑
7.18(m,1h),7.14(d,j=7.2hz,2h),6.96(d,j=8.4hz,1h),5.58
‑
5.40(m,2h),4.90(宽峰s.,1h),4.65
(宽峰s.,1h),3.94(宽峰s.,1h),3.70(d,j=14.0hz,1h),3.36(宽峰s.,3h),3.22(宽峰s.,3h),3.16
‑
2.90(m,2h),2.39
‑
2.25(m,1h),1.79
‑
1.56(m,6h),1.31(s,9h)。
[0120]
lc/ms(方法7):保留时间2.72min,97%uv纯度(220nm),m/z质量观测值469.2。
[0121]
sfc(方法5):保留时间2.36min,>96%uv纯度(220nm)。
[0122]
实例7:新戊酰氧基甲基2a和2b的制备
[0123][0124]
以类似于1a1和1b1的方式制备(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓碘化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓碘化物(2a2;x=i
‑
和2b2;x=i
‑
)以及(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(2a2;x=cl
‑
和2b2;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(2;6.00g)和在乙腈(42ml)中的新戊酸碘甲酯(3b;8.00g),以提供约5g粗碘化物和约4g(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(2a2;x=cl
‑
和2b2;x=cl
‑
)的hplc纯化(方法3)混合物。将该混合物通过手性sfc(方法6)分离。如之前使用方法11和3纯化两种产物以提供两种产物:
[0125]
第一洗脱异构体:1.00g的(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(2a2;x=cl
‑
)或(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(2b2;x=cl
‑
)。
[0126]1h nmr:400mhz甲醇
‑
d
4 7.71(宽峰s,1h),7.36(dd,j=2.0,8.1hz,1h),6.97(d,j=7.9hz,1h),5.61(宽峰s,1h),5.52(d,j=8.8hz,1h),5.00
‑
4.89(m,1h),4.68(s,1h),3.85(宽峰s,2h),3.54
‑
3.31(m,3h),3.28
‑
3.16(m,1h),2.99(宽峰dd,j=10.3,13.8hz,1h),2.43
‑
2.28(m,1h),1.71(宽峰s,6h),1.32(s,9h)。
[0127]
lc/ms(方法7):保留时间2.72min,98.9%uv纯度(220nm),m/z质量观测值477.3。
[0128]
sfc(方法5):保留时间1.79min,>99%uv纯度(220nm)。
[0129]
第二洗脱异构体:0.72g的(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(2b2;x=cl
‑
)或(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)
‑1‑
((新戊酰氧基)甲基)哌嗪
‑1‑
鎓氯化物(2a2;x=cl
‑
)。
[0130]1h nmr:400mhz甲醇
‑
d
4 7.44(d,j=2.2hz,1h),7.25(dd,j=2.0,8.1hz,1h),6.94(d,j=8.3hz,1h),5.52
‑
5.45(m,1h),5.45
‑
5.39(m,1h),4.58(dd,j=3.9,7.9hz,1h),4.46(t,j=7.7hz,1h),3.74(宽峰s,1h),3.53(宽峰d,j=12.7hz,1h),3.09
‑
2.74(m,4h),2.63(宽峰s,1h),2.21
‑
2.04(m,1h),1.69
‑
1.48(m,6h),1.38
‑
1.24(m,9h)。
[0131]
lc/ms(方法7):保留时间2.72min,97.6%uv纯度(220nm),m/z质量观测值477.3。
[0132]
sfc(方法5):保留时间2.1min,>97%uv纯度(220nm)。
[0133]
实例8:十二烷酰氧基甲基1a和1b的制备
[0134][0135]
以类似于1a1和1b1的方式制备(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓碘化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓碘化物(1a3;x=i
‑
和1b3;x=i
‑
)以及(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1a3;x=cl
‑
和1b3;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(1;7.00g)和在乙腈(49ml)中的月桂酸碘甲酯(3c;16.8g)以提供约5.5g粗碘化物。将该材料通过反相柱色谱(方法18)纯化,随后进行离子交换(方法11)以提供3.2g(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1a3;x=cl
‑
和1b3;x=cl
‑
)的混合物。
[0136]1h nmr:400mhz甲醇
‑
d
4 7.45(s,1h),7.31
‑
7.23(m,3h),7.23
‑
7.17(m,1h),7.13
‑
7.07(m,2h),6.94(d,j=8.3hz,1h),5.52(宽峰s,1h),5.40(dd,j=3.9,8.8hz,1h),4.64
‑
4.55(m,1h),4.47(宽峰t,j=7.2hz,1h),3.81
‑
3.64(m,1h),3.56(宽峰d,j=14.5hz,1h),3.11(d,j=7.5hz,3h),3.04
‑
2.91(m,2h),2.86
‑
2.75(m,2h),2.57(dt,j=1.8,7.5hz,2h),2.19
‑
2.06(m,1h),1.71
‑
1.56(m,8h),1.29(宽峰s,17h),0.93
‑
0.84(m,3h)。
[0137]
lc/ms(方法13):保留时间2.66min,99.3%uv纯度(220nm),m/z质量观测值567.3。
[0138]
实例9:十二烷酰氧基甲基2a和2b的制备
[0139][0140]
以类似于1a3和1b3的方式制备(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓碘化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓碘化物(2a3;x=i
‑
和2b3;x=i
‑
)以及(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a3;x=cl
‑
和2b3;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(2;5.60g)和在乙腈(39ml)中的月桂酸碘甲酯(3c;10.5g),以提供约5g粗碘化物和3.1g(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((十二烷酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a3;x=cl
‑
和2b3;x=cl
‑
)的混合物。
[0141]1h nmr:400mhz甲醇
‑
d
4 7.49(宽峰s,1h),7.27(d,j=8.3hz,1h),6.94(d,j=8.3hz,1h),5.52(宽峰s,1h),5.46
‑
5.36(m,1h),4.69
‑
4.58(m,1h),4.50(宽峰t,j=7.5hz,1h),3.86
‑
3.66(m,1h),3.61(宽峰s,1h),3.16
‑
2.79(m,4h),2.57(dt,j=1.3,7.5hz,2h),2.23
‑
2.07(m,1h),1.76
‑
1.51(m,8h),1.47
‑
1.19(m,17h),0.95
‑
0.83(m,3h)。
[0142]
lc/ms(方法13):保留时间2.68min,98.9%uv纯度(220nm),m/z质量观测值575.4。
[0143]
实例10:环己基乙酰氧基甲基1a和1b的制备
[0144][0145]
以类似于1a1和1b1的方式制备(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓碘化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓碘化物(1a4;x=i
‑
和1b4;x=i
‑
)以及(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1a4;x=cl
‑
和1b4;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪(1,6.00g)和在乙腈(42ml)中的2
‑
环己基乙酸碘甲酯(3d;11.9g),以提供约8g粗碘化物和3.2g(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1a4;x=cl
‑
和1b4;x=cl
‑
)的hplc纯化混合物(方法8)。将该混合物通过手性sfc(方法9)分离。如之前使用方法11和8纯化两种产物以提供两种产物:
[0146]
第一洗脱异构体:1.00g的(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1a4;x=cl
‑
)或(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1b4;x=cl
‑
)。
[0147]1h nmr:400mhz甲醇
‑
d
4 7.57(宽峰s,1h),7.32
‑
7.26(m,3h),7.25
‑
7.19(m,1h),7.15
‑
7.09(m,2h),6.95(d,j=8.3hz,1h),5.56(宽峰s,1h),5.45(d,j=8.8hz,1h),4.74(宽峰s,1h),4.57(宽峰t,j=7.5hz,1h),3.84
‑
3.60(m,2h),3.27
‑
2.77(m,8h),2.46(d,j=7.0hz,2h),2.23(td,j=7.2,14.5hz,1h),1.99
‑
1.47(m,12h),1.39
‑
1.13(m,3h),1.12
‑
0.96(m,2h)
[0148]
lc/ms(方法14):保留时间2.85min,97.4%uv纯度(220nm),m/z质量观测值509.3。
[0149]
sfc(方法10):保留时间2.45min,>99%uv纯度(220nm)。
[0150]
第二洗脱异构体:0.33g的(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1b4;x=cl
‑
)或(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
1,2,2
‑
三甲基哌嗪
‑1‑
鎓氯化物(1a4;x=cl
‑
)。
[0151]1h nmr:400mhz甲醇
‑
d
4 7.51(s,1h),7.33
‑
7.26(m,3h),7.25
‑
7.19(m,1h),7.14
‑
7.09(m,2h),6.96(d,j=8.8hz,1h),5.56
‑
5.47(m,1h),5.46
‑
5.39(m,1h),4.72
‑
4.64(m,
1h),4.52(宽峰t,j=7.7hz,1h),3.79(宽峰s,1h),3.62(宽峰s,1h),3.21
‑
2.77(m,8h),2.46(d,j=6.8hz,2h),2.26
‑
2.12(m,1h),1.85
‑
1.55(m,12h),1.43
‑
1.16(m,3h),1.12
‑
0.97(m,2h)。
[0152]
lc/ms(方法15):保留时间3.66min,97.7%uv纯度(220nm),m/z质量观测值509.2。
[0153]
sfc(方法10):保留时间6.41min,>97%uv纯度(220nm)。
[0154]
实例11:环己基乙酰氧基甲基2a和2b的制备
[0155][0156]
以类似于1a1和1b1的方式制备(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓碘化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓碘化物(2a4;x=i
‑
和2b4;x=i
‑
)以及(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a4;x=cl
‑
和2b4;x=cl
‑
)的混合物。合成始自4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪(2;6.00g)和在乙腈(42ml)中的2
‑
环己基乙酸碘甲酯(3d;11.7g),以提供约7.8g粗碘化物和6g(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物和(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a4;x=cl
‑
和2b4;x=cl
‑
)的hplc纯化(方法8)混合物。将该混合物通过手性sfc(方法16)分离。
[0157]
如之前使用方法11和8纯化两种产物以提供两种产物:
[0158]
第一洗脱异构体:1.10g的(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a4;x=cl
‑
)或(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2b4;x=cl
‑
)。
[0159]1h nmr:400mhz甲醇
‑
d
4 7.50(宽峰s,1h),7.30(宽峰d,j=8.2hz,1h),6.97(d,j=8.2hz,1h),5.54(宽峰s,1h),5.49
‑
5.41(m,1h),4.66(宽峰s,1h),4.58
‑
4.48(m,1h),3.73
(宽峰s,1h),3.62(宽峰s,1h),3.21
‑
2.74(m,5h),2.47(d,j=6.8hz,2h),2.26
‑
2.12(m,1h),1.92
‑
1.57(m,12h),1.38
‑
1.17(m,3h),1.14
‑
0.98(m,2h)。
[0160]
lc/ms(方法14):保留时间2.68min,96.6%uv纯度(220nm),m/z质量观测值517.2。
[0161]
sfc(方法10):保留时间2.33min,>97%uv纯度(220nm)。
[0162]
第二洗脱异构体:1.10g的(s)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2b4;x=cl
‑
)或(r)
‑4‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑1‑
((2
‑
环己基乙酰氧基)甲基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪
‑1‑
鎓氯化物(2a4;x=cl
‑
)。
[0163]1h nmr:400mhz甲醇
‑
d
4 7.48(宽峰s,1h),7.28(d,j=8.2hz,1h),6.95(d,j=8.2hz,1h),5.49(宽峰s,1h),5.44
‑
5.38(m,1h),4.64(宽峰d,j=4.0hz,1h),4.49(宽峰s,1h),3.83
‑
3.70(m,1h),3.58(宽峰d,j=13.2hz,1h),3.16
‑
2.63(m,5h),2.45(d,j=6.8hz,2h),2.16(宽峰dd,j=6.9,10.7hz,1h),1.91
‑
1.51(m,12h),1.43
‑
1.16(m,3h),1.13
‑
0.95(m,2h)。
[0164]
lc/ms(方法7):保留时间2.99min,99.4%uv纯度(220nm),m/z质量观测值517.3。
[0165]
sfc(方法10):保留时间4.87min,>97%uv纯度(220nm)。
[0166]
实例12:血浆稳定性测定
[0167]
将冷冻的人血浆(储存在
‑
80℃)在水浴中解冻,随后在3200
×
g下离心5min以去除碎片。然后测量上清液的ph值并且通过添加1%磷酸或1n氢氧化钠调节至7.4
±
0.1。
[0168]
对于每种测试化合物,将2μl给药溶液(对于测试化合物为50μm并且对于阳性对照为100μm;溴丙胺太林)加标(spiked)到98μl空白血浆中以达到1μm测试化合物和2μm阳性对照的最终浓度。
[0169]
将测试化合物和阳性对照在6个不同时间点(0小时、0.5小时、1小时、2小时、4小时和6小时)在37℃下的水浴中与人血浆一式两份孵育(最终dmso浓度<1%)。在每个相应的时间点,通过添加适当体积的淬灭溶液以停止反应而终止孵育。
[0170]
然后,将血浆样品短暂涡旋,并且随后在3200
×
g下离心20min。将上清液转移至96孔板中,并且在lc
‑
ms/ms分析之前用200μl超纯水以1:2的比率稀释。分析物/内标物的峰面积比(par)用于半定量地确定测试化合物和对照化合物的浓度。报告在各个时间点残余的测试化合物相对于0分钟样品的百分比。
[0171]
在确定母体药物和对照的体外消除常数(ke)时,将分析物/内标物峰面积比用以下公式转化成残余百分比:
[0172][0173]
通过代谢物药物的相对于t0(绝对)的分析物/内标物峰面积比计算代谢物的形成百分比并且转化成百分比。
[0174]
由分析物/内标物峰面积比对时间的对数线性图计算母体药物和对照化合物的半衰期(t1/2)(t1/2=0.693/ke)。
[0175]
将化合物1a2、1b2、2a2、以及2b2如上所描述在人血浆中孵育,并且由分别形成15%
‑
35%的4
‑
((1r,3s)
‑6‑
氯
‑3‑
苯基
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
1,2,2
‑
三甲基哌嗪和4
‑
((1r,3s)
‑6‑
氯
‑3‑
(苯基
‑
d5)
‑
2,3
‑
二氢
‑
1h
‑
茚
‑1‑
基)
‑
2,2
‑
二甲基
‑1‑
(甲基
‑
d3)哌嗪来确
定半衰期为0.5
‑
1.5h。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1