一种左布比卡因中间体化合物的制作方法
1.本发明属于医药合成技术领域,具体涉及一种左布比卡因中间体化合物。
背景技术:
2.左布比卡因(levoaupivacaine),化学名为(2s)-1-丁基-n-(2,6-二甲基苯基)-2-哌啶甲酰胺,是布比卡因的s型光学异构体,由英国cellechchiroscience公司研制,2000年3月在美国首次上市,经动物实验及临床应用表明其药理学特性与布比卡因相仿,麻醉效能相近,毒副作用较小是一种新型长效的酰胺类局部麻醉药。左布比卡因主要应用于外科和产科手术的局部麻醉及用于术后疼痛,但在大剂量使用或静脉注射时,布比卡因消旋体具有较大的心脏毒性和神经毒性,其毒性主要来自右旋布比卡因,左布比卡因临床安全性更高,具有良好的应用价值。因此,越来越多的组织和机构对单一异构体左布比卡因的合成越来越感兴趣。其cas号为27262-47-1,化学结构如下所示:
[0003][0004]
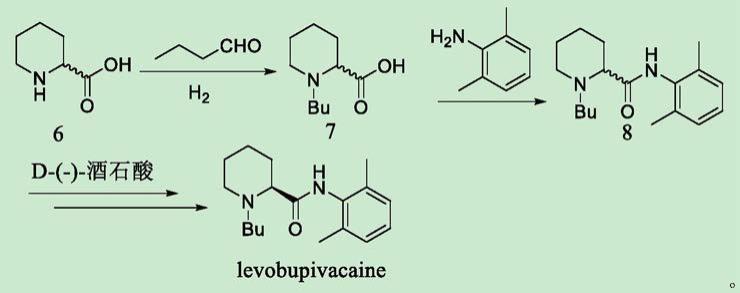
目前,文献报道的盐酸左布比卡因主要制备方法是以左哌啶酰胺和1-溴丁烷为起始物料,以碳酸钾等无机碱为催化剂,以n,n-二甲基甲酰胺,经高温、长时间反应得到左布比卡因碱基,再用盐酸成盐得到盐酸左布比卡,如中国专利申请cn107098849报道了以2-哌啶甲酸为原料,经亚胺还原得到化合物6,化合物6与2,6-二甲基苯胺在活化剂下缩合得布比卡因消旋体,最后经酒石酸拆分得到左布比卡因。该路线需要进行化学拆分,经处理得到单一异构体,产物收率与异构纯度较低,需要多次拆分,会延长反应操作时间,增加溶剂用量和生产成本,合成路线如下:
[0005][0006]
中国专利申请cn105418489a、us5777124、wo9612700、ep14337821报道了以一种2-哌啶甲酸为原料,经hcl酸化成盐得化合物9,再经氯代试剂二氯亚砜得到化合物10,化合物10与2,6-二甲基苯胺缩合得到化合物11,化合物11与1-溴丁烷反应得到布比卡因消旋体,经拆分剂拆分和后处理得到左布比卡因。该路线中应用到多种酸源,对溶剂的含水量有一
定的要求,对仪器腐蚀度较大;步骤较长,涉及拆分过程,产物的收率与纯度较低。合成路线如下:
[0007][0008]
专利wo2009089842a以2-吡啶甲酸盐酸盐为原料,与草酰氯反应得到2-哌啶甲酰氯(化合13),与2,6-二甲基甲酰胺缩合得到化合物14,氢气还原得到化合物11,与1-溴丁烷反应得到化合物8,经拆分以及后处理得到左布比卡因。该路线相对较长,氢气还原需要特殊反应装置,此外涉及产物的化学拆分收率与异构纯度较低,合成路线如下:
[0009][0010]
文献(tetrahedron letters,2005,46(1):19-21)报道了一种不对称合成左布比卡因的方法,化合物15与化合物16合成化合物17,化合物17与叠氮化钠反应,随后氢化还原得到化合物18,二苯甲酮亚胺与化合物18反应得到化合物19,化合物19在立体选择性催化剂条件下与1-碘-4氯丁烷反应生成单一异构体化合物20,化合物20与氰基硼氢化钠氢化关环反应得到化合物21,化合物21与氯化钯在高压下脱去二苯甲基得到化合物5,化合物5与溴丁烷反应得到左布比卡因。该路线反应步骤长,应用到一些昂贵的催化剂,会极大增加生产成本;此外叠氮化钠极不稳定,易爆炸,不利于安全生产,合成路线如下:
[0011][0012]
综上所述,在已经被报道的制备左布比卡因的技术方法中,主要存在的问题为:
[0013]
1、哌啶2位立体选择性引入手性基团需要一定的立体选择性催化剂,通常这些催化剂比较昂贵,反应产物的异构纯度相对较低;在手性基团的构建过程中,需要引入或使用特殊的氨源,像叠氮化钠,亚硝酸钠等,这些化合物性状不稳定,需严格控制反应过程,安全性较差,不利于大生产的操作;
[0014]
2、现有路线中,应用较多的酸源,草酸,酰氯,盐酸等对设备腐蚀性较大,对人体和环境也会造成危害;
[0015]
3、哌啶的羧酸基团与2,6-二甲基苯胺进行缩合时,需要一定的催化剂,增加苯胺的反应活性,相比脂肪胺,收率相对较低,反应效率差。
[0016]
综上所述,寻找一条反应条件温和,操作过程简便,产品收率高、纯度高,生产成本低,原子利用率高的适合工业化生产左布比卡因的制备方法仍是目前需要解决的问题。
技术实现要素:
[0017]
针对目前现有左布比卡因制备技术存在的问题,本发明提供了一种左布比卡因中间体化合物iv并提供了该化合物的制备方法,以及利用该化合物合成左布比卡因的方法。该制备左布比卡因方法反应条件温和,操作过程简便,生产成本低,原料利用率高,所制得的目标产品具有较高的纯度、收率。
[0018]
本发明的具体技术方案如下:
[0019]
一种如式i
ⅴ
所示的左布比卡因中间体化合物:
[0020][0021]
化合物iv制备方法包括如下步骤:将化合物ii、化合物iii及碱加入有机溶剂中,控温搅拌至反应结束,反应液减压浓缩后经重结晶得中间体iv,合成路线如下:
[0022][0023]
优选的,所述的碱选自三乙胺、吡啶、n-甲基吗啉、碳酸氢钠、碳酸钾、碳酸钠中的一种或其组合,特别优选碳酸钠;
[0024]
优选的,所述的化合物ii与碱的投料摩尔比为1:1.2~2.0,特别优选1:1.5;
[0025]
优选的,所述的化合物ii与化合物iii的投料摩尔比为1:1.2~2.0,特别优选1:1.4;
[0026]
优选的,所述重结晶溶剂选自甲基叔丁基醚、异丙醚、乙醚、二氯甲烷、四氢呋喃中的一种或其组合,特别优选异丙醚;
[0027]
优选的,所述重结晶温度为-10℃~5℃,特别优选-5℃~0℃;
[0028]
优选的,所述的有机溶剂选自n,n-二甲基甲酰胺、二甲基亚砜、甲苯、二甲苯中的一种或其组合,特别优选n,n-二甲基甲酰胺;
[0029]
优选的,所述的反应温度为60℃~100℃,特别优选90℃~95℃;
[0030]
化合物i
ⅴ
用于制备左布比卡因的用途。
[0031]
化合物i
ⅴ
用于制备左布比卡因的用途制备方法包括如下步骤:将化合iv、2.6-二甲基苯胺加入有机溶剂中,加入碱、催化剂及配体,惰性气体保护下加入氯仿及一水合氢氧化铯,控温搅拌反应得左布比卡因,制备路线如下:
[0032][0033]
优选的,所述的催化剂是铜类催化剂,选自碘化亚铜、氧化亚铜、氯化亚铜、醋酸铜、氯化铜、氧化铜中的一种或其组合,特别优选氯化亚铜。
[0034]
优选的,所述配体选自1,10-菲啰啉、三苯基膦、2,2
’-
联吡啶、双(2-二苯基磷苯基)醚、三环己基膦中的一种或其组合,特别优选1,10-菲啰啉。
[0035]
优选的,所述化合物iv、催化剂、配体的投料摩尔比1:0.05~0.15:0.05~0.15,特别优选1:0.1:0.1。
[0036]
优选的,所述的碱选自碳酸铯、碳酸钾、碳酸钠、氟化铯、碳酸氢钠中的一种或其组合,特别优选碳酸铯。
[0037]
优选的,所述的化合物iv、2,6-二甲基苯胺、碱的投料摩尔比为1:1:2~2.0:1.5~2.5,特别优选1:1.4:2.0。
[0038]
优选的,中所述的化合物iv、氯仿、一水合氢氧化铯投料摩尔比为1:2.0~5.0:8.0~12.0,特别优选1:3.0:10.0。
[0039]
优选的,所述的有机溶剂选自1.4-二氧六环、甲苯、二甲苯,n,n-二甲基甲酰胺、二甲基亚砜中的一种或其组合,特别优选甲苯。
[0040]
优选的,所述的反应温度为80~120℃,最优温度在100~105℃。
[0041]
本发明取得的技术效果是:
[0042]
1、提供了一种新的左布比卡因中间体化合物iv,在制备左布比卡因后续取代反应中无较大杂质产生。
[0043]
2、利用该新中间体得到的左布比卡因,避免了羧基与胺基低效的缩合,避免拆分,并且所得产品纯度也较高。
[0044]
3、该中间体合成路线简单,时间短,收率以及纯度较高。
具体实施方式
[0045]
下面通过实施例来进一步说明本发明,应该正确理解的是:本发明的实施例仅仅是用于说明本发明,而不是对本发明的限制,所以,在本发明的方法前提下对本发明的简单改进均属本发明要求保护的范围。
[0046]
新化合物结构表征:
[0047][0048]
化合物iv的高分辨质谱:esi-hrms:m/z=176.1130[m+h]
+
;1h-nmr(400mhz,cdcl3):δ1.2-1.6(m.3h),1.65-1.94(m,4h),2.05-2.53(m,2h),2.74-3.32(m,4h),3.45-3.66(m.1h).7.20(m.2h).7.48(m.2h);
13
c-nmr(100mhz,cdcl3):δ23.9,24.8,28.12,29.3,31.7,32.0,32.3,34.0,34.3.5.76,54.3,63.6,64.7,65.4,67.2,126.5.126.7,129.0,129.1,131.1,133.4.
[0049]
本发明采用hplc测定化合物i的纯度,有关色谱条件:
[0050]
色谱柱:agela technologies promosil c18,(4.6mm
×
250mm,5.0μm);
[0051]
流动相:0.02mol/l磷酸缓冲盐(磷酸二氢钾2.72g与氢氧化钠0.75g,加水1000ml使溶解,调节ph值为8.0)-乙腈(35:65);
[0052]
检测波长:210nm;
[0053]
实施例1
[0054]
将化合物ii(12.00g,0.10mol),化合物iii(19.18g,0.14mol),碳酸钠(15.90g,0.15mol)加入n,n-二甲基甲酰胺(30ml),升温至90~95℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用异丙醚(20ml)-5℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率96.5%,hplc纯度99.89%。
[0055]
实施例2
[0056]
将化合物ii(12.00g,0.10mol),化合物iii(16.44g,0.12mol),三乙胺(15.18g,0.15mol)加入乙腈(30ml),升温至60~65℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用异丙醚(20ml)在-5℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率92.6%,hplc纯度99.82%。
[0057]
实施例3
[0058]
将化合物ii(12.0g,0.10mol),化合物iii(13.70g,0.10mol),吡啶(11.87g,0.15mol)加入n,n-二甲基甲酰胺(30ml),升温至55~60℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用四氢呋喃(20ml)-10℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率86.6%,hplc纯度99.78%。
[0059]
实施例4
[0060]
将化合物ii(12.0g,0.10mol),化合物iii(27.40g,0.20mol),n-甲基吗啉(15.17g,0.15mol)加入乙腈(30ml),升温至95~100℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用异丙醚(20ml)0℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率93.7%,hplc纯度99.75%。
[0061]
实施例5
[0062]
将化合物ii(12.0g,0.10mol),化合物iii(34.25g,0.25mol),碳酸氢钠(12.60g,0.15mol)加入二甲亚砜(30ml),升温至100~105℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用异丙醚(20ml)5℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率87.9%,hplc纯度99.71%。
[0063]
实施例6
[0064]
将化合物ii(12.0g,0.10mol),化合物iii(19.18g,0.14mol),碳酸钠(12.72g,0.12mol)加入甲苯(30ml),升温至90~95℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用乙醚(20ml)0℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率92.9%,hplc纯度99.83%。
[0065]
实施例7
[0066]
将化合物ii(12.0g,0.10mol),化合物iii(19.18g,0.14mol),碳酸钠(10.60g,0.1mol)加入对二甲苯(30ml),升温至90~95℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用二氯甲烷(20ml)0℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率87.2%,hplc纯度99.71%。
[0067]
实施例8
[0068]
将化合物ii(12.0g,0.10mol),化合物iii(19.18g,0.14mol),碳酸钠(21.20g,0.20mol)加入间二甲苯(30ml),升温至90~95℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用四氢呋喃(20ml)-3℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率93.2%,hplc纯度99.79%。
[0069]
实施例9
[0070]
将化合物ii(12.0g,0.10mol),化合物iii(19.18g,0.14mol),碳酸钠(23.32g,0.22mol)加入n,n-二甲基甲酰胺(30ml),升温至90~95℃反应5h后,反应液降温至室温后,减压浓缩至干,将所得固体用异丙醚(20ml)-7℃下重结晶后,过滤,将所得滤饼40℃减压真空干燥后即为化合物iv,收率88.4%,hplc纯度99.63%。
[0071]
左布比卡因的制备
[0072]
实施例10
[0073]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(13.33g,0.11mol)加入甲苯(100ml),氯化亚铜(0.79g,10mol%),1,10-菲啰啉(1.59g,10mol%),碳酸铯(52.13g,0.16mol),氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,
0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率98.3%,hplc纯度为99.85%。
[0074]
实施例11
[0075]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(12.11g,0.10mol)加入甲苯(100ml),碘化亚铜(1.52g,10mol%),三苯基膦(2.10g,10mol%),碳酸铯(52.13g,0.16mol),氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率94.6%,hplc纯度为99.81%。
[0076]
实施例12
[0077]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(9.70g,0.08mol)加入甲苯(100ml),氧化亚铜(1.14g,10mol%),2,2
’-
联吡啶(1.25g,10mol%),碳酸铯(52.13g,0.16mol),氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率86.7%,hplc纯度为99.75%。
[0078]
实施例13
[0079]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(19.39g,0.16mol)加入甲苯(100ml),醋酸铜(1.60g,10mol%),2,2
’-
联吡啶(1.25g,10mol%),碳酸钾(22.11g,0.16mol),氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率94.8%,hplc纯度为99.72%。
[0080]
实施例14
[0081]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(24.24g,0.2mol)加入甲苯(100ml),醋酸铜(1.60g,10mol%),2,2
’-
联吡啶(1.25g,10mol%),碳酸钾(22.11g,0.16mol),氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率87.2%,hplc纯度为99.67%。
[0082]
实施例15
[0083]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入1.4-二氧六环(100ml),氯化亚铜(0.79g,10mol%),1,10-菲啰啉(1.59g,10mol%),碳酸铯(52.13g,0.16mol),氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至80~85℃反应24h后,反应液过滤,将1.4-二氧六环浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率94.2%,hplc纯度为99.78%。
[0084]
实施例16
[0085]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入n,n-二甲基甲酰胺(100ml),氯化铜(二水)(1.36g,10mol%),1,10-菲啰啉(1.59g,10mol%),碳酸铯(32.59g,0.1mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至115~120℃反应24h后,反应液过滤,将n,n-二甲基甲酰胺浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率88.2%,hplc纯度为99.72%。
[0086]
实施例17
[0087]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入对二甲苯(100ml),氯化亚铜(0.79g,10mol%),1,10-菲啰啉(1.59g,10mol%),碳酸铯(65.16g,0.20mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至120~125℃反应24h后,反应液过滤,将对二甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率93.6%,hplc纯度为99.75%。
[0088]
实施例18
[0089]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入二甲亚砜(100ml),氧化铜(0.64g,10mol%),1,10-菲啰啉(1.59g,10mol%),碳酸铯(71.68g,0.22mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至75~80℃反应24h后,反应液过滤,将二甲亚砜浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率87.7%,hplc纯度为99.61%。
[0090]
实施例20
[0091]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入甲苯(100ml),氯化亚铜(0.40g,5mol%),双(2-二苯基磷苯基)醚(2.15g,5mol%),碳酸钠(16.96g,0.16mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率94.5%,hplc纯度为99.78%。
[0092]
实施例21
[0093]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入甲苯(100ml),氯化亚铜(0.32g,4mol%),三环己基膦(0.90g,4mol%),氟化铯(24.30g,0.16mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率89.2%,hplc纯度为99.72%。
[0094]
实施例22
[0095]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入甲苯(100ml),氯化亚铜(1.19g,15mol%),三环己基膦(3.37g,15mol%),氟化铯(24.30g,0.16mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml
打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率93.2%,hplc纯度为99.71%。
[0096]
实施例23
[0097]
将化合物iv(14.00g,0.08mol),2,6-二甲基苯胺(14.54g,0.12mol)加入甲苯(100ml),氯化亚铜(1.68g,17mol%),2,2
’-
联吡啶(2.00g,16mol%),碳酸氢钠(13.44g,0.16mol)氮气保护下,加入氯仿(28.65g,0.24mol)和一水合氢氧化铯(134.34g,0.80mol);升温至100~105℃反应24h后,反应液过滤,将甲苯浓缩至干,向浓缩液中加入纯化水200ml打浆,过滤,所得滤饼50℃减压真空干燥后即为左布比卡因,收率87.4%,hplc纯度为99.66%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1