索马鲁肽侧链的汇聚式液相合成法的制作方法
本发明属于多肽合成领域,具体涉及一种治疗二型糖尿病的多肽类药物索马鲁肽的侧链的汇聚式液相合成方法。
背景技术:
据统计,2017年全球糖尿病患者人数已达4.25亿。由于不可治愈性以及人口老龄化加速,预计2045年全球糖尿病患者人数将达到6.29亿。索马鲁肽(胰高血糖素样肽-1(glp-1))注射剂和口服制剂(用于控制血糖降低心血管事件风险概率)的上市无疑将对糖尿病患者带来福音。索马鲁肽的分子序列为:
his-aib-glu-gly-thr-phe-thr-ser-asp-val-ser-ser-tyr-leu-glu-gly-gln-ala-ala-lys(aeea-aee-γ-glu-octadecanedioicacidmono-tert-butylester)-glu-phe-ile-ala-trp-leu-val-arg-gly-arg-gly-oh
第26位lys所连侧链可以看成以下四单元组成:2分子8-氨基-3,6-二氧杂辛酸、一分子谷氨酸和一分子十八烷二酸单叔丁酯,侧链的存在可以增强索马鲁肽药物与蛋白的结合,从而减低肾脏清除率。目前工业上常用直线式固相合成法的方式合成索马鲁肽及其侧链,由于索马鲁肽侧链原料相对昂贵,开发新的合成方式,降低合成成本和周期将是当务之急。
本发明采用液相合成法汇聚式合成上述lys所连接的侧链1。
技术实现要素:
本发明的目的在于提供一种索马鲁肽侧链的汇聚式液相合成法,该方法能够降低液相合成的成本,缩短反应周期,提高反应收率,具有工业化应用前景。
本发明所述索马鲁肽侧链的结构式如下所示:
本发明的技术方案如下:
一种索马鲁肽侧链1的合成方法,所述方法包括如下步骤:
(1)将原料二甘醇胺2的端氨基用r1保护,接着与α卤代酯发生亲核取代反应,并通过酯水解一锅制得化合物4;
所述r1为fmoc、alloc、boc、pmb、cbz、trt、tos、mtt、mmt、bom、sem或mem,优选fmoc、trt或boc,更优选trt或boc;r1保护所用的反应溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选乙醇或甲醇,更优选甲醇;
所述α卤代酯为溴乙酸乙酯或溴乙酸苄酯,优选溴乙酸乙酯;与α卤代酯发生亲核取代反应所用的反应溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选四氢呋喃或乙腈,更优选四氢呋喃;
酯水解在无机碱的作用下进行,所述无机碱选自氢氧化钠、氢氧化锂、碳酸钾、碳酸钠、氢氧化钙、氢氧化钡中的一种或几种任意比例的混合物,优选氢氧化钠;所述无机碱与原料二甘醇胺2的物质的量之比为0.5~4:1,优选0.5~2:1,更优选2:1;
(2)化合物4游离的羧基用r2保护,并且脱除r1保护基,得到化合物6;
所述r2为bn、pfp、me、allyl、t-bu、pmb、mem或tbs,优选bn或me,更优选bn;
脱除r1保护基在酸试剂的作用下进行,所述酸试剂为盐酸、醋酸或三氟乙酸,优选三氟乙酸;脱除r1保护基所用的反应溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选甲醇或二氯甲烷,更优选二氯甲烷;特别优选的,脱除r1保护基在三氟乙酸:二氯甲烷体积比1:1~2(优选1:1)的体系中进行;
(3)芴甲氧羰基-l-谷氨酸1-叔丁酯7与化合物6经缩合反应,得到化合物8;
所述缩合反应在缩合剂的作用下进行,所述缩合剂选自dic、dcc、hbtu、pybop、bop、hatu、tbtu、dic/hobt、dcc/dmap、edci/dmap、edci/hobt、hatu/hobt中的任意单一缩合剂或者复合缩合剂,优选edci/dmap、edci/hobt或hatu,更优选edci/hobt,并且edci和hobt的物质的量之比为1:0.5~2,优选1:1;所述缩合反应的温度在20~70℃,优选20~40℃,更优选30℃;所述缩合反应的溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选1,2-二氯乙烷或二氯甲烷,更优选二氯甲烷;
(4)将化合物8的芴甲氧羰基脱除,再与18-(叔丁氧基)-18氧代十八烷酸进行酰胺化缩合偶联反应,得到化合物11;
芴甲氧羰基的脱除在有机碱的作用下进行,所述有机碱选自二乙胺、n,n-二异丙基乙胺、三乙胺、哌啶、咪唑、吡啶或dbu,优选三乙胺、dbu或二乙胺,更优选二乙胺;芴甲氧羰基脱除所用的反应溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选乙腈或甲醇,更优选乙腈;
与18-(叔丁氧基)-18氧代十八烷酸进行酰胺化缩合偶联反应在缩合剂的作用下进行,所述缩合剂选自dic、dcc、hbtu、pybop、bop、hatu、tbtu、dic/hobt、dcc/dmap、edci/dmap、edci/hobt、hatu/hobt中的任意单一缩合剂或者复合缩合剂,优选edci/dmap、edci/hobt或hatu,更优选edci/hobt,并且edci和hobt的物质的量之比为1:0.5~2,优选1:1;缩合偶联反应的溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选1,2-二氯乙烷或二氯甲烷,更优选二氯甲烷;缩合偶联反应的温度在20~70℃,优选20~40℃,更优选30℃;
(5)化合物11脱除r2保护基,再与化合物6进行缩合偶联反应,得到化合物13;
r2保护基的脱除在无机碱的作用下进行,所述无机碱选自氢氧化钾、氢氧化钡、氢氧化钙、氢氧化铝、氢氧化锂、氢氧化镁或氢氧化锌,优选氢氧化钾或氢氧化锂,更优选氢氧化锂,推荐无机碱以水溶液的形式投料,例如0.5~2m(优选1m)氢氧化锂水溶液;脱除r2保护基所用的反应溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选乙醇或甲醇,更优选甲醇;
与化合物6进行缩合偶联反应在缩合剂的作用下进行,所述缩合剂选自dic、dcc、hbtu、pybop、bop、hatu、tbtu、dic/hobt、dcc/dmap、edci/dmap、edci/hobt、hatu/hobt中的任意单一缩合剂或者复合缩合剂,优选edci/dmap、edci/hobt或hatu,更优选edci/hobt,并且edci和hobt的物质的量之比为1:0.5~2,优选1:1;所述缩合偶联反应的溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选1,2-二氯乙烷或二氯甲烷,更优选二氯甲烷;所述缩合偶联反应的温度在20~70℃,优选20~40℃,更优选30℃;
(6)化合物13脱除r2保护基,得到目标产物链1;
r2保护基的脱除在无机碱的作用下进行,所述无机碱选自氢氧化钾、氢氧化钡、氢氧化钙、氢氧化铝、氢氧化锂、氢氧化镁或氢氧化锌,优选氢氧化钾或氢氧化锂,更优选氢氧化锂,推荐无机碱以水溶液的形式投料,例如0.5~2m(优选1m)氢氧化锂水溶液;脱除r2保护基所用的反应溶剂选自甲醇、乙醇、乙酸乙酯、二氯甲烷、1,2-二氯乙烷、dmf、dmso、四氢呋喃、乙腈、n-甲基吡咯烷酮中的一种或几种任意比例的混合溶剂,优选乙醇或甲醇,更优选甲醇。
本发明最终合成的产物可直接与索马鲁肽lys残基上游离的氨基发生缩合反应从而连接至索马鲁肽主链上。
本发明的优点在于:使用汇聚式合成的方法降低反应成本,缩短了反应时间。合成过程有效、可控、低成本高产率,可适用于放大生产。避免了易燃且昂贵的钯碳试剂的使用。
本发明中的缩写含义如下:
his:组氨酸
aib:2-甲基丙氨酸
glu:谷氨酸
gly:甘氨酸
thr:苏氨酸
phe:苯丙氨酸
thr:苏氨酸
ser:丝氨酸
asp:天冬氨酸
val:缬氨酸
leu:亮氨酸
gln:谷氨酰胺
ala:丙氨酸
lys:赖氨酸
fmoc:芴甲氧羰基
alloc:烯丙氧羰基
boc:叔丁氧羰基
pmb:对甲氧基苄基
trt:三苯甲基
tos:对甲苯磺酰基
mtt:4-甲基-三苯甲基
mmt:4-甲氧基三苯甲基
sem:三甲基硅乙氧基甲基
mem:2-甲氧基乙氧基甲基
edci:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐
hobt:1-羟基苯并三唑
hatu:o-(7-氮杂苯并三唑-1-基)-n,n,n′,n′-四甲基脲
dic:n,n'-二异丙基碳二亚胺
dcc:二环己基碳二亚胺
hbtu:苯并三唑-1-四甲基六氟磷酸酯
dmap:4-二甲氨基吡啶
pybop:1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐
bop:苯并三唑-1-三(三甲氨基)-三氟磷酸酯
tbtu:o-(ih-苯并三唑-1-基)-n,n,n',n'-四甲基异脲四氟化硼
附图说明
图1为本发明实施例2的合成路线图。
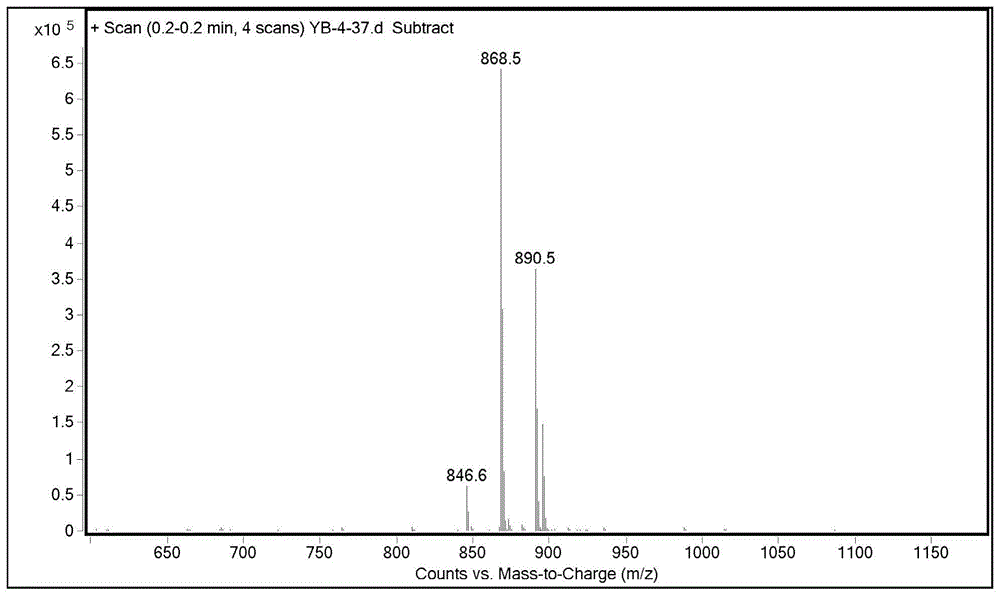
图2为化合物1的ms图。
图3为化合物1的红外谱图。
具体实施方式
下面结合具体实施例对本发明作进一步的说明,但本发明的保护范围并不仅限于此。
实施例1
r1优选为trt,r2优选为bn
实施例1-1:
取500ml圆底烧瓶,将10ml二甘醇胺2(100mmol)溶于二氯甲烷溶液中,0℃下缓慢滴加14ml三乙胺(1eq),搅拌30min后缓慢滴加trt-cl(1.2eq),撤除冰浴,室温下反应1h。减压蒸馏除去溶剂,残留物使用四氢呋喃重新溶解,冰浴下加入28g碳酸钾,搅拌30min后滴加溴乙酸苄酯(1.5eq),撤除冰浴,将反应液升温至50℃搅拌过夜。减压蒸馏除去溶剂,乙酸乙酯萃取两次,饱和食盐水洗涤,无水硫酸钠干燥。减压蒸馏得目标产物。收率为90%。
实施例1-2:化合物6的制备。
将上述产物溶解在二氯甲烷溶液中,加入三氟乙酸:二氯甲烷(v:v)=1:1,tlc监测反应,减压蒸馏除去三氟乙酸得到黄色油状液体6待用。
实施例1-3:化合物8的制备。
取50ml圆底烧瓶,将3g芴甲氧羰基-l-谷氨酸1-叔丁酯(7mmol)使用二氯甲烷溶解,分别加入3.6ml的dipea,1.6gedci和1.1ghobt,搅拌30min后将化合物6(1.1eq)加入反应液,30℃下搅拌,tlc监测反应。减压蒸馏除去溶剂,残留物用乙酸乙酯重新溶解,分别使用10%的柠檬酸(50ml×2),饱和碳酸氢钠溶液(50ml×2)萃取,饱和食盐水洗,收集有机层,无水硫酸钠干燥减压蒸馏除去溶剂,柱层析纯化(ch2cl2:meoh=20:1)得黄色色油状体8。收率82%。
实施例1-4:化合物9的制备
将3.3g化合物8溶于22ml乙腈溶液中,逐滴加入11.6ml二乙胺,室温下搅拌2h,tlc监测反应。旋除溶剂,柱层析纯化(ch2cl2:meoh=10:1)得黄色油状物9,收率86%。
实施例1-5:化合物11的制备。
取50ml圆底烧瓶,使用二氯甲烷将1.11g的18-(叔丁氧基)-18氧代十八烷酸溶解,分别加入1.5ml的dipea,691mg的edci和486mghobt,30℃下搅拌30min后加入1.57g化合物9,tlc监测反应。减压蒸馏除去溶剂,残留物用乙酸乙酯重新溶解,10%的柠檬酸,饱和碳酸氢钠溶液萃取,饱和食盐水洗,收集有机层,无水硫酸钠干燥。减压蒸馏除去溶剂,柱层析纯化(ch2cl2:meoh=30:1)得黄色油状体11。收率89%。
实施例1-6:化合物12的制备。
取一圆底烧瓶,将791g(1mmol)化合物11溶于10ml甲醇溶液中,向反应液中逐滴加入1mol/l的lioh水溶液4ml,室温下搅拌2h,减压蒸馏除去溶剂,通过柱层析纯化(ch2cl2:meoh:hcooh=30:1:1),得化合物12。收率77%。
实施例1-7:化合物13的制备。
取25ml圆底烧瓶,将上述化合物12(0.77mmol,539mg)溶于5ml的二氯甲烷中,依次加入dipea(3eq,0.4ml),edci/hobt(1.2eq),搅拌30min后加入化合物6(1.2eq),tlc监测反应。反应完毕,减压蒸馏除去溶剂,残留物用乙酸乙酯重新溶解,10%的柠檬酸,饱和碳酸氢钠溶液萃取,饱和食盐水洗,收集有机层,使用无水硫酸钠干燥。减压蒸馏除去溶剂得油状液体13,收率82%。
实施例1-8:化合物1的制备。
取25ml圆底烧瓶,将化合物13溶解于2mlmeoh,向反应液中逐滴加入1mol/l的lioh水溶液,室温下搅拌2h,减压蒸馏除去溶剂,通过柱层析纯化(ch2cl2:meoh=5:1),得最终产物1。收率90%。
实施例2:
具体实验路线可参照图1
r1优选为boc,r2优选为bn。
实施例2-1:化合物4的制备。
一锅法制备:取250ml圆底烧瓶,将10ml的2-(2-氨基乙氧基)乙醇溶于150ml的乙醇中,冰浴条件下缓慢滴加23ml二碳酸二叔丁酯,滴加完毕撤除冰浴,室温下反应2h,减压蒸馏除去乙醇溶剂;残留物使用thf重新溶解,保持0~-10℃投入nah(1.2-1.5eq),搅拌30min,将16.6ml的溴乙酸乙酯3缓慢滴入反应液,室温下搅拌过夜,tlc监测反应。向反应体系加入meoh,使meoh:thf(v:v)=1:1,称取8gnaoh加入反应液,加热回流反应2h。减压蒸馏除去溶剂,残留物使用水重新溶解,乙酸乙酯萃取两次,收集水层,调成ph1~3,乙酸乙酯萃取两次,收集有机层,减压蒸馏得黄色油状液体4。收率为82%。
实施例2-2:化合物6的制备。
(1)冰浴条件下,向上述所述原料4(1-1.2eq)和三乙胺(1-1.2eq)的二氯甲烷(100ml)溶液中缓慢滴加氯甲酸苄酯5(100mmol,12.9ml),搅拌5min后向反应液加入dmap(0.1-0.3eq),室温下过夜反应,tlc监测反应(ch2cl2:ea=10:1)。反应完毕,减压蒸馏除去溶剂,残留物用乙酸乙酯重新溶解,依次使用10%的柠檬酸(75ml×2),饱和碳酸氢钠溶液(75ml×2)萃取,饱和食盐水(100ml)洗,收集有机层,使用无水硫酸钠干燥。减压蒸馏除去溶剂得无色油状液体。收率63%。
(2)将上述无色油状液体溶解在二氯甲烷溶液中,使用三氟乙酸:二氯甲烷(v:v)=1:1脱boc保护,减压蒸馏除去三氟乙酸得到黄色油状液体6待用。
后续制备得到最终产物1的步骤与实施例1中相同。
以上具体实施例用来补充和说明本发明的可行性,在不背离本发明的精神和范围的情况下可以作出许多其它的更改和修改。因此,所附权利要求范围还应包含在本发明的基础上的变化和修改。
- 还没有人留言评论。精彩留言会获得点赞!