一种罗沙司他的制备方法与流程
1、技术领域
本发明属于药物化学技术领域,具体涉及一种罗沙司他的制备方法。
2、
背景技术:
罗沙司他(roxadustat,fg-4592)最初由美国fibrogen(珐博进)开发。2006年4月,astellas与fibrogen达成许可协议,获得了罗沙司他在欧洲、独联体、中东及南非的开发权。2013年7月31日,astrazeneca(阿斯利康)和fibrogen达成战略合作协议,在美国、中国以及上述astellas许可地区之外主要市场的开发权。
2018年12月18日,由珐博进中国和阿斯利康中国合作开发的罗沙司他首先在中国获准上市,用于治疗正在接受透析治疗的患者因慢性肾脏病(ckd)引起的贫血。上市制剂为胶囊,规格为20mg、50mg,商品名为爱瑞卓。罗沙司他的结构如下式所示。
中国发明专利申请cn1816527a、cn102718708a、cn102977015a、cn102977016a、cn103145616a公开了一种以4-苯氧基邻苯二甲腈为原料,经11步反应制备罗沙司他的方法,反应路线如下所示。该方法所用试剂三溴氧磷、碘甲烷、溴化苄以及氯甲酸酯类毒性较大;反应时间较长:多步操作反应时间大于18h,有的高达3.5d;反应条件较苛刻:如第二步210℃熔融搅拌反应,甲基化反应-78℃超低温进行,最后一步氢化加压安全隐患较大;中间体多步经柱层析纯化,步骤较长,成本较高,不适合工业化生产。
中国发明专利申请cn103435546a、cn106083719a公开了一种以5-溴苯酞为原料,经9步反应合成罗沙司他的方法,反应路线如下所示。该工艺第一步醚化反应,5-溴苯酞易开环生成较多杂质;工艺所用试剂乙酸酐易制毒、属于管制品,第四步使用对甲苯磺酰基甘氨酸,可能引入磺酸酯类基因毒性杂质;工艺中使用重金属,如溴化亚铜、钯碳,成品中增加元素检测;两步加压步骤:氢化加压安全隐患较大,最后一步酯的氨解反应较难进行,需要特殊设备,不适合工业化生产,工艺成本较高。
中国发明专利申请cn107954931a公开了一种以-羟基-7-苯氧基异喹啉-3-甲酸甲酯为原料,经溴代、偶联水解、缩合以及酯水解合成罗沙司他的方法,反应路线如下所示。该工艺每一步反应都需要回流反应6h或者过夜反应,整个工艺反应时间较长,能耗较大;第一步溴代反应以甲醇为溶剂,容易产生甲氧基取代杂质;第二步反应容易产生水解杂质,反应体系较杂,难以精制,中间体纯度控制较困难,产率较低。
中国发明专利申请cn107602466a公开了一种以3-硝基苄胺为原料,经10步反应合成罗沙司他的方法,反应路线如下所示。该工艺起始原料不易得,价格较贵;使用的氯乙酸中等毒性,氯甲烷致癌具有基因毒性,高锰酸钾氧化性较强,盐酸、硫酸、液溴以及氯乙酸均为管制品,上述试剂具有一定安全隐患;第一步反应温度高达150℃,第九步反应高达220℃;氯甲烷、氨气钢瓶存放具有较大的安全隐患,不适合工业化生产。
因此,研究一种反应原料易得、反应原料毒性低、合成路线简单、反应时间短、操作简便、成本低、不污染环境且适合工业化大生产的罗沙司他的制备方法是本领域亟待解决的问题。
3、
技术实现要素:
本发明的目的是解决现有技术中存在的问题,提供了一种起始原料便宜易得,反应原料毒性低,合成路线简短,反应周期短,不污染环境,操作简便避免苛刻的反应条件,操作及后处理简便,收率及产品纯度较高,各步中间体均为固体且易于纯化,生产成本较低,适合工业化大生产,符合绿色化学原则的罗沙司他的新的制备方法,以克服现有技术问题。
为了达到上述的目的,本发明提供了一种制备罗沙司他的方法,包括以下步骤:
(1)式i化合物与卤化试剂发生取代反应,生成式ii化合物;
(2)式ii化合物在有机溶剂和碱的存在下,发生水解反应,生成式iii化合物;
(3)式iii化合物与甘氨酸酯盐酸盐在缩合剂的存在下,发生缩合反应,生成式v化合物;
(4)式v化合物在催化剂的催化下,与甲基化试剂在有机溶剂和无机碱的水溶液的混合溶液中发生甲基化反应并同时进行水解反应,得到罗沙司他;
其中,x为卤素;r1和r2为c1-6烷基。
步骤(1)中,所述的卤化试剂选自1,3-二氯-5,5-二甲基海因、1,3-二溴-5,5-二甲基海因、1,3-二碘-5,5-二甲基海因、n-氯代丁二酰亚胺、n-溴代丁二酰亚胺、n-碘代丁二酰亚胺、溴素、单质碘中的一种或者几种;优选地,为n-溴代丁二酰亚胺。
步骤(1)中,反应温度为10℃~70℃;优选地为25℃。
步骤(1)中,反应时间为0~5h小时。
步骤(1)中,式i化合物与卤化试剂的反应摩尔比为1:(1~2)。
步骤(2)中,所述的碱选自氢氧化钠、氢氧化钾、氢氧化锂、碳酸钠、碳酸钾、碳酸氢钠、碳酸铯中的一种或几种;优选地为氢氧化钠。
步骤(2)中,反应温度为50℃~120℃。
步骤(2)中,反应时间为0~5h小时。
步骤(3)中,反应温度为0℃~50℃。
步骤(3)中,反应时间为1~5h小时。
步骤(4)中,所述的甲基化试剂选自甲基硼酸、甲基硼酸异丙酯、三甲基硼、四甲基锡、甲基溴化镁、甲基氯化镁中的一种或几种;优选地为甲基硼酸。
步骤(4)中,所述有机溶剂选自甲基叔丁基醚、乙二醇二甲醚、乙二醇甲醚、乙二醇二乙醚、乙二醇乙醚、甲苯、四氢呋喃、2-甲基四氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜、1,4-二氧六环、乙腈中的一种或几种;优选地为乙二醇甲醚。
步骤(4)中,所述的催化剂为钯类催化剂,选自pdcl2、pd2(dba)3、pd(oac)2、pd(dppf)cl2、pd(pph3)4、pd(pph3)2cl2、pd(pph3)2(oac)2的一种或几种;优选地,为pd(pph3)4。
步骤(4)中,所述的无机碱的水溶液选自碳酸氢钠、乙酸钾、碳酸钾、醋酸钠、碳酸氢钾、碳酸铯、碳酸钠、乙醇钠、甲醇钠、磷酸钾、磷酸钠中的一种或几种的水溶液;优选地,为磷酸钾的水溶液。
步骤(4)中,反应温度为70℃~130℃;优选地为80℃~110℃。
步骤(4)中,反应时间为6~36h;优选地为12~24h。
进一步地,本发明所述的步骤(3),还可以任选地包括下述的反应步骤:
(a)式iii化合物与氯化亚砜发生取代反应,生成式iv化合物;
(b)式iv化合物与甘氨酸酯盐酸盐在缚酸剂的存在下,发生取代反应,生成式v化合物;
其中,x为卤素,r2为c1-6烷基。
步骤(a)中,反应温度为25℃~80℃。
步骤(a)中,反应时间为0~5h小时。
步骤(a)中,式iii化合物与氯化亚砜的反应摩尔比为1:(1~5)。
步骤(b)中,所述的缩合试剂选自n,n-羰基二咪唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、二环己基碳二亚胺、n,n'-二异丙基碳二亚胺、1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)、1-羟基苯并三氮唑、1-羟基-7-偶氮苯并三氮唑、o-(7-氮杂苯并三唑-1-基)-n,n,n′,n′-四甲基脲六氟磷酸酯、o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸酯中的一种或几种,优选为1h-苯并三唑-1-基氧三吡咯烷基六氟磷酸盐(pybop)。
步骤(b)中,反应温度为15℃~50℃。
步骤(b)中,反应时间为1~5h小时。
本发明所述的卤素选自氟、氯、溴、碘;优选地,氯、溴、碘;最优选地为溴。
本发明所述的c1-6烷基是指从含有1至6个碳原子的c1-6烃化合物中除去一个氢原子所得到的一价基团,所述的烃化物可以是脂肪族或脂环族的或是其组合形式,并且可以是饱和的、部分饱和的或完全饱和的。饱和的直链c1-6烷基的例子包括但不限于甲基、乙基、正丙基、正丁基和正戊基。饱和的支链c1-6烷基的例子包括但不限于异丙基、异丁基、仲丁基、叔丁基和新戊基。饱和的脂环族(碳环)的c1-6烷基的例子包括但不限于的基团如环丙基、环丁基、环戊基、环己基、甲基环丙基、二甲基环丙基、甲基环丁基、二甲基环丁基、甲基环戊基和环丙基甲基。优选地为甲基、叔丁基。
本发明的有益效果为:
(1)本发明提供了一种新的用于制备罗沙司他的方法。
(2)本发明制备罗沙司他的方法,起始反应原料4-羟基-7-苯氧基异喹啉-3-甲酸甲酯易得,可直接购买获得,且毒性低,非管制物品。
(3)本发明制备罗沙司他的方法,合成路线短,共4步反应。
(4)本发明制备罗沙司他的方法,反应时间短,约20h-22h。
(5)本发明制备罗沙司他的方法,收率高,每步反应的收率大于83.20%,总收率大于71.51%、高达80.82%。
(6)本发明制备罗沙司他的方法,后处理采用重结晶的方法,不需过柱纯化,操作简单。
(7)与对比例1相比,用本发明方法制备获得的中间体1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯杂质少,lc-ms谱图未见甲氧基取代的杂质峰,中间体易于控制。
(8)与对比例2相比,用本发明方法制备获得的1-甲基-4-羟基-7-苯氧基异喹啉-3-甲酸即罗沙司他杂质少,纯度高于98%,lc-ms谱图未见1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯水解的杂质峰。
(9)本发明提供的制备方法,反应条件温和,操作简便,采用的溶剂均为常规溶剂,无加压反应,无安全隐患,易于控制,工艺成本低,适合工业化生产。
4、附图说明
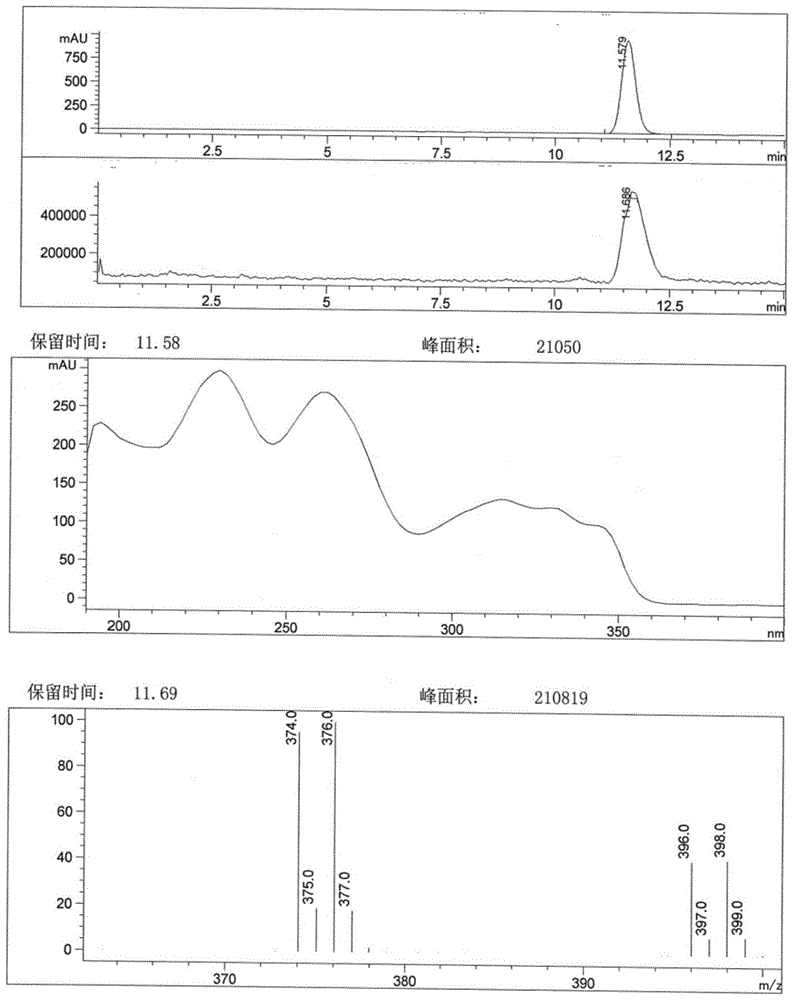
图1为实施例1制备的1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯的lc-ms谱图。
图2为实施例8制备的(4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酰基)甘氨酸的lc-ms谱图。
图3为对比例1制备的1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯的lc-ms谱图。
图4为对比例2制备的1-甲基-4-羟基-7-苯氧基异喹啉-3-甲酸的lc-ms谱图。
5、具体实施方式
下面详细描述本发明的具体实施例,需要说明的是下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
实施例1:1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯
将4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(50.00g,169.45mmol)加入至500ml四氢呋喃中,然后加入nbs(31.67g,177.93mmol)以及aibn(1.39g,8.47mmol)。加毕,25℃反应1h。tlc分析(石油醚-乙酸乙酯=3:2),反应完全。后处理:减压浓缩除去大部分溶剂,然后加入500ml甲醇升温至回流打浆0.5h,降温、过滤,约用50ml甲醇洗涤滤饼,晾干得到60.60g类白色固体,产率95.64%,lc-ms谱图见图1,谱图显示未见明显杂质峰。
1h-nmr(600mhz,dmso-d6):δ3.00(3h,s),7.27(2h,dd,j=7.2,1.2hz),7.34(1h,t,j=7.2hz),7.41(1h,d,j=2.4hz),7.54(2h,td,j=7.2,1.2hz),7.69(1h,dd,j=9.0,2.4hz),8.37(1h,d,j=9.0hz),11.54(1h,s)。
实施例2:1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸
将1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(71.00g,189.74mmol)加入至300ml乙醇中,然后加入氢氧化钠(22.71g,569.22mmol)和300ml水的混合液。加毕,升温至回流反应约1h。tlc分析(石油醚-乙酸乙酯=5:1),反应完全。后处理:降温,减压浓缩除去部分溶剂,后加入500ml水,2m盐酸调节ph至2-3,搅拌析晶0.5h后,过滤,干燥得到67.70g淡红色固体,有关物质99.31%,产率99.06%。
1h-nmr(600mhz,dmso-d6):δ7.27(2h,dd,j=7.2,1.2hz),7.34(1h,t,j=7.2hz),7.42(1h,d,j=2.4hz),7.54(2h,td,j=7.2,1.2hz),7.68(1h,dd,j=9.0,2.4hz),8.36(1h,d,j=9.0hz),13.08(约2h,brs)。
实施例3:(1-溴-4-羟基-7-苯氧基异喹啉-3-羰基)甘氨酸甲酯
将1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸(20.00g,55.53mmol)加入至250ml二氯甲烷中,然后加入甘氨酸甲酯盐酸盐(10.46g,83.30mmol)以及pybop(43.35g,83.30mmol),最后加入三乙胺(22.48g,222.12mmol)。加毕,控温25℃反应约2h。tlc分析(dcm-meoh(10:1)+3dhac),反应基本完全。后处理:向反应体系加入120ml水,分液,二氯甲烷萃取2次(200ml×2),合并有机相,无水硫酸镁干燥,活性炭脱色,过滤,减压浓缩得到75.02g类白色固体。
精制:向上述粗品中加入400ml异丙醇,升温回流打浆0.5h,降温,过滤,约用30ml异丙醇洗涤滤饼,干燥得到24.00g类白色固体,产率约100%。
1h-nmr(600mhz,dmso-d6):δ3.69(3h,s),4.13(2h,d,j=6.6hz),7.27(2h,dd,j=7.2,1.2hz),7.34(1h,tt,j=7.2,1.2hz),7.46(1h,d,j=2.4hz),7.54(2h,td,j=7.2,1.2hz),7.69(1h,dd,j=9.0,2.4hz),8.36(1h,d,j=9.0hz),9.30(1h,brs),13.47(约1h,brs)。
实施例4:(1-溴-4-羟基-7-苯氧基异喹啉-3-羰基)甘氨酸叔丁酯
将1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸(10.00g,27.77mmol)加入至150ml二氯甲烷中,然后加入甘氨酸叔丁酯盐酸盐(6.98g,41.66mmol)以及pybop(21.68g,41.66mmol),最后加入三乙胺(11.24g,111.08mmol)。加毕,控温25℃反应约2h。tlc分析(dcm-meoh(10:1)+3dhac),反应基本完全。后处理:向反应体系加入120ml水和150ml二氯甲烷,分液,二氯甲烷萃取2次(100ml×2),合并有机相,无水硫酸镁干燥,活性炭脱色,过滤,减压浓缩得到38.01g类白色固体。
精制:向上述粗品中加入200ml异丙醇,升温回流打浆0.5h,降温,过滤,约用15ml异丙醇洗涤滤饼,干燥,得到12.53g类白色固体,产率约95.35%。
1h-nmr(600mhz,dmso-d6):δ1.44(9h,s),4.00(2h,d,j=6.0hz),7.27(2h,dd,j=7.2,1.2hz),7.34(1h,tt,j=7.2,1.2hz),7.47(1h,d,j=2.4hz),7.54(2h,td,j=7.2,1.2hz),7.70(1h,dd,j=9.0,2.4hz),8.36(1h,d,j=9.0hz),9.16(1h,t,j=6.0hz),13.61(1h,s)。
实施例5:1-溴-4-羟基-7-苯氧基异喹啉-3-甲酰氯
将1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸(5.0g,13.88mmol)加入至50ml二氯甲烷中,然后滴入氯化亚砜(4.13g,34.70mmol)。加毕,回流反应约2h。tlc分析(dcm-meoh(10:1)+3dhac),反应完全。后处理:减压浓缩除去溶剂得到5.26g淡黄色固体,产率约100%。留存备用,直接用于下一步反应。
实施例6:(1-溴-4-羟基-7-苯氧基异喹啉-3-羰基)甘氨酸甲酯
将甘氨酸甲酯盐酸盐(2.61g,20.82mmol)加入至26ml二氯甲烷中,然后加入三乙胺(5.62g,55.52mmol)。降温,控温至25℃以下,滴加1-溴-4-羟基-7-苯氧基异喹啉-3-甲酰氯(5.26g,13.88mmol)和50ml二氯甲烷混合液。加毕,缓慢升至室温反应约2h。tlc分析(dcm-meoh(10:1),反应基本完全。后处理:向反应体系加入40ml水和40ml二氯甲烷,分液,二氯甲烷萃取1次(50ml×1),合并有机相,无水硫酸镁干燥,过滤,减压浓缩得粗品。
精制:将上述粗品加入至33ml异丙醇中,升温至回流约0.5h,降温析晶,过滤,约用5ml异丙醇洗涤滤饼,干燥得到5.51g类白色固体,产率约92.06%。
1h-nmr(600mhz,dmso-d6):δ3.69(3h,s),4.13(2h,d,j=6.6hz),7.27(2h,dd,j=7.2,1.2hz),7.34(1h,tt,j=7.2,1.2hz),7.46(1h,d,j=2.4hz),7.54(2h,td,j=7.2,1.2hz),7.69(1h,dd,j=9.0,2.4hz),8.36(1h,d,j=9.0hz),9.30(1h,brs),13.47(约1h,brs)。
实施例7:(1-溴-4-羟基-7-苯氧基异喹啉-3-羰基)甘氨酸叔丁酯
将甘氨酸叔丁酯盐酸盐(3.49g,20.82mmol)加入至35ml二氯甲烷中,然后加入三乙胺(5.62g,55.52mmol)。降温,控温至25℃以下,滴加1-溴-4-羟基-7-苯氧基异喹啉-3-甲酰氯(5.26g,13.88mmol)和50ml二氯甲烷混合液。加毕,缓慢升至室温反应约2h。tlc分析(dcm-meoh(10:1),反应基本完全。后处理:向反应体系加入50ml水和50ml二氯甲烷,分液,二氯甲烷萃取1次(50ml×1),合并有机相,无水硫酸镁干燥,过滤,减压浓缩得粗品。
精制:将上述粗品加入至33ml异丙醇中,升温至回流约0.5h,降温析晶,过滤,约用5ml异丙醇洗涤滤饼,干燥得到5.96g类白色固体,产率约90.72%。
1h-nmr(600mhz,dmso-d6):δ1.44(9h,s),4.00(2h,d,j=6.0hz),7.27(2h,dd,j=7.2,1.2hz),7.34(1h,tt,j=7.2,1.2hz),7.47(1h,d,j=2.4hz),7.54(2h,td,j=7.2,1.2hz),7.70(1h,dd,j=9.0,2.4hz),8.36(1h,d,j=9.0hz),9.16(1h,t,j=6.0hz),13.61(1h,s)。
实施例8:(4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酰基)甘氨酸
将(1-溴-4-羟基-7-苯氧基异喹啉-3-羰基)甘氨酸甲酯(10.00g,23.19mmol)加入至100ml乙二醇甲醚中,然后加入甲基硼酸(8.33g,139.14mmol),磷酸钾(18.21g,85.80mmol),25ml水以及四(三苯基膦)钯(1.61g,1.39mmol)。加毕,氮气置换3次,升温至100℃反应约16h。tlc分析(dcm-meoh(10:1)+2dhac),反应完全。后处理:降温,向反应体系中加入48ml乙二醇甲醚和25ml水,过滤,2m盐酸调节滤液ph至2-3,搅拌析晶约0.5h,过滤,约用25ml水洗涤滤饼,晾干得到8.20g淡黄色固体,产率约100%。
精制:将上述粗品加入至82ml乙醇中,升温至80℃回流搅拌约10min。降温,过滤,约用5ml乙醇洗涤滤饼,晾干得到6.97g淡黄色固体,有关物质98.73%,产率85.31%。lc-ms谱图见图2,谱图显示未见明显杂质峰。
1h-nmr(600mhz,dmso-d6):δ2.71(3h,s),4.05(2h,d,j=6.0hz),7.19(2h,d,j=7.2hz),7.26(1h,t,j=7.2hz),7.49(2h,t,j=7.2hz),7.54(1h,td,j=9.0,2.4hz),7.62(1h,d,j=2.4hz),8.30(1h,d,j=9.0hz),9.10(1h,t,j=6.0hz),12.68(约1h,brs),14.33(1h,brs)。
实施例9:(4-羟基-1-甲基-7-苯氧基异喹啉-3-甲酰基)甘氨酸
将(1-溴-4-羟基-7-苯氧基异喹啉-3-羰基)甘氨酸叔丁酯(10.00g,21.13mmol)加入至100ml乙二醇甲醚中,然后加入甲基硼酸(7.59g,126.78mmol),磷酸钾(16.60g,78.18mmol),25ml水以及四(三苯基膦)钯(1.47g,1.27mmol)。加毕,氮气置换3次,升温至100℃反应约16h。tlc分析(dcm-meoh(10:1)+2dhac),反应完全。后处理:降温,向反应体系中加入80ml乙二醇甲醚和25ml水,过滤,2m盐酸调节滤液ph至2-3,搅拌析晶约0.5h,过滤,约用25ml水洗涤滤饼,晾干得到7.50g淡黄色固体,产率约100%。
精制:将上述粗品加入至75ml乙醇中,升温至80℃回流搅拌约10min。降温,过滤,约用10ml乙醇洗涤滤饼,晾干得到6.19g淡黄色固体,有关物质98.29%,产率83.20%。
1h-nmr(600mhz,dmso-d6):δ2.71(3h,s),4.05(2h,d,j=6.0hz),7.19(2h,d,j=7.2hz),7.26(1h,t,j=7.2hz),7.49(2h,t,j=7.2hz),7.54(1h,td,j=9.0,2.4hz),7.62(1h,d,j=2.4hz),8.30(1h,d,j=9.0hz),9.10(1h,t,j=6.0hz),12.68(约1h,brs),14.33(1h,brs)。
对比例1:1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯
将4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(0.50g,1.69mmol)加入至7.5ml甲醇中,降温、控温10℃以下,加入1,3-二溴-5,5-二甲基海因(0.27g,0.93mmol)。加毕,升温至回流反应约6h。tlc分析(石油醚-乙酸乙酯=3:2),反应基本完全。后处理:缓慢降温至5℃左右,过滤,约用3.5ml甲醇洗涤滤饼,晾干得到0.40g类白色固体,批号:19111903。lc-ms显示推测为甲氧基取代杂质,无溴原子同位素峰,非目标产物,见图3。
对比例2:1-甲基-4-羟基-7-苯氧基异喹啉-3-甲酸
将1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯(6.00g,16.03mmol)加入至105ml乙二醇甲醚中,然后加入甲基硼酸(8.33g,139.14mmol),磷酸钾(12.52g,58.99mmol),20ml水以及四(三苯基膦)钯(0.56g,0.48mmol)。加毕,氮气置换3次,升温至100℃过夜反应。tlc分析(dcm-meoh(10:1)+2dhac),反应完全。后处理:降温,15ml乙二醇甲醚和10ml水依次洗涤滤饼,2m盐酸调节滤液ph至2-3,搅拌析晶约0.5h,过滤,约用25ml水洗涤滤饼,晾干得到4.20g淡黄色固体,产率约81.08%。lc-ms显示产品纯度较差,含有较多1-溴-4-羟基-7-苯氧基异喹啉-3-甲酸甲酯的水解杂质,见图4。
- 还没有人留言评论。精彩留言会获得点赞!