一种硝苯地平的制备方法与流程
本申请涉及有机药物合成
技术领域:
,具体而言,涉及一种硝苯地平的制备方法。
背景技术:
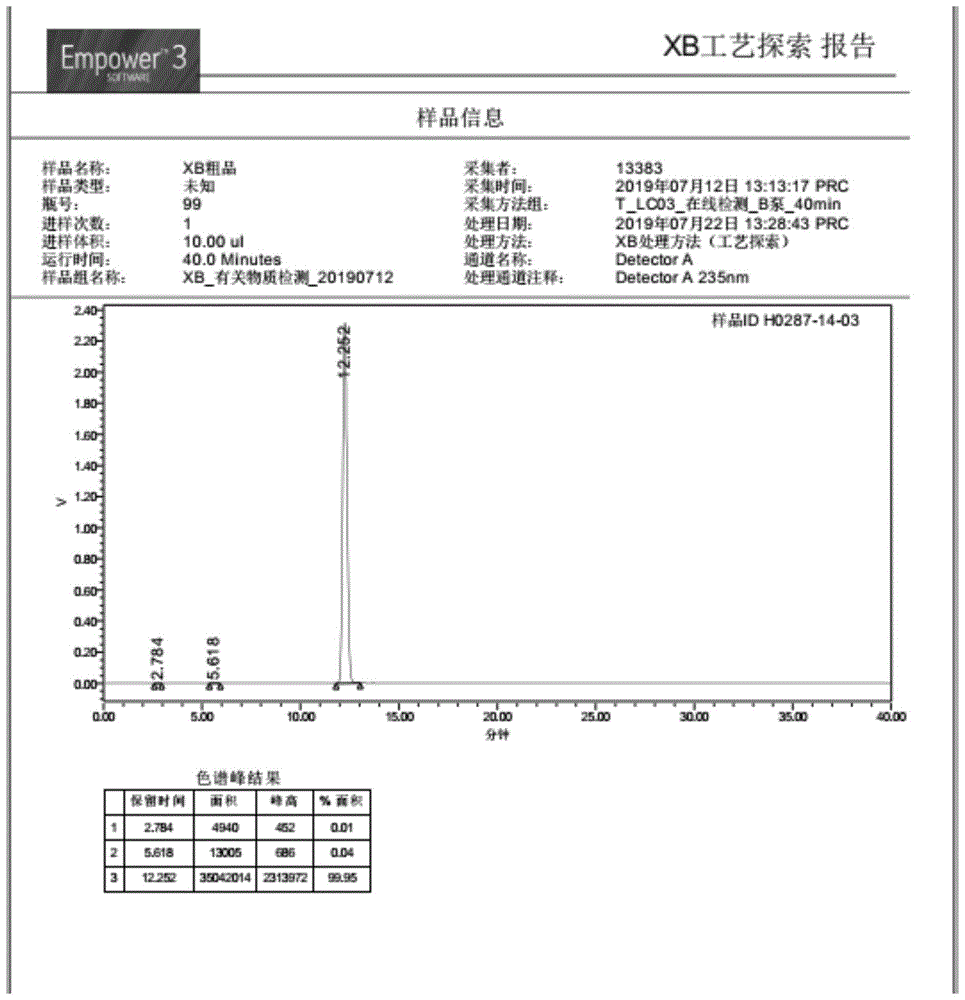
:硝苯地平(nifedipine)是目前临床上应用最广泛的二氢吡啶类降压药物之一。化学名为:2,6-二甲基-4-(2-硝基苯基-)-1,4二氢-3,5-吡啶二甲酸二甲酯,化学结构式如下:由于硝苯地平的分子结构中二氢吡啶环上共有两对左右对称结构的边链—二甲酸二甲酯和二甲基,因而硝苯地平的常用合成路线为一步合成法。即经典的hantzsch二氢吡啶合成法的一个具体应用。合成路线如下:在实际应用中,由于氨水碱性偏强,会发生较多的副反应,从而生成较多的副产物,故多使用碱性偏弱的碳酸氢铵、醋酸铵替代氨水,可以使硝苯地平的产物纯度提高,但提高不明显。于是,后续研究者采用3-氨基巴豆酸甲酯代替上述氨化试剂,并采取错时加料的方法,希望最大程度地减少副产物的产生。如cn1190422c先将邻硝基苯甲醛与乙酰乙酸甲酯发生knovennogel缩合反应,使生成邻硝基亚苄基乙酰乙酸甲酯中间体,再将3-氨基巴豆酸甲酯加入此反应液中,在同一溶液中,使3-氨基巴豆酸甲酯与硝苯地平中间体发生环合反应生成目标产物硝苯地平。反应式具体如下:这种错时加料、“两步反应一锅烩”的工艺相较于上述“一步反应一锅烩”的工艺,产品纯度有明显提高。如中国药典(2015年版)中载明的两个氧化性杂质含量均能控制在药典规定限度以下,但残余的起始原料—邻硝基苯甲醛(基因毒性杂质)含量仍然有偏高的情况。原因是:虽然采取了错时加料措施,却未将第一步反应产物硝苯地平中间体(邻硝基亚苄基乙酰乙酸甲酯)自第一步反应液中沉淀分离出来,则第一步反应中未作用的剩余起始原料(邻硝基苯甲醛)及其他更多的未知工艺杂质继续被留在下一步反应的反应液中。而且残留的邻硝基苯甲醛很可能与3-氨基巴豆酸甲酯发生如下副反应:此杂质在且在后续精制过程中较不易清除,并成为成品中最常见的工艺杂质之一。尤其是继续留在硝苯地平粗品中的邻硝基苯甲醛很难在后续精制过程中被充分清除,或即使清除充分,却会导致不应有的产品损失,从而降低收率。为了充分降低起始原料邻硝基苯甲醛在终产品中的残留量,并避免其与第二步反应试剂—3-氨基巴豆酸甲酯发生不应有的副反应,将硝苯地平现有的“一步反应一锅烩”或“两步反应一锅烩”方法改变为“两步反应两锅烩”的新工艺,将成为合成药用级硝苯地平的一种新途径。技术实现要素:本申请的目的在于提供一种硝苯地平的制备方法,以便使硝苯地平粗品中基因毒性杂质(邻硝基苯甲醛)含量达标,从而避免粗品精制过程中不应有的产品损失,同时又能较大幅度地缩短反应时间,提高生产效率。本申请实施例提供一种硝苯地平的制备方法,在含氮杂环羧酸催化剂的作用下,中间体邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯在c1-c4的低级脂族醇类溶剂中发生加成反应,得到硝苯地平。使用上述含氮杂环羧酸催化剂,使邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯反应生成硝苯地平,在缩短反应时间的同时,降低了粗品中基因毒性杂质(邻硝基苯甲醛)残留量,从而可省去为降低基因毒性杂质残留量而设置的繁复的后续纯化过程,达到简化工艺、提高收率的双重效果。附图说明为了更清楚地说明本申请实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本申请的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图也属于本申请的保护范围。图1为本申请实施例1技术条件下的加成反应终点反应液的液相色谱图;图2为实施例8技术条件下的加成反应终点反应液的液相色谱图;图3为实施例11技术条件下的加成反应终点反应液的液相色谱图;图4为对比例1技术条件下的加成反应终点反应液的液相色谱图;图5为实施例8所得的硝苯地平粗品经用乙醇一次重结晶后所得硝苯地平精制样品的液相色谱图。具体实施方式为使本申请实施例的目的、技术方案和优点更加清楚,下面将对本申请实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。硝苯地平的制备方法,在含氮杂环羧酸催化剂的作用下,邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯在c1-c4低级脂肪族醇类溶剂中发生加成反应,得到硝苯地平。将上述含氮杂环羧酸催化剂用于邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯加成反应后,可以显著降低硝苯地平粗品和成品中的杂质(尤其是特殊单杂)含量,从而获得纯度更高的硝苯地平粗品和成品。同时明显缩短反应时间,提高反应效率。其中,含氮杂环羧酸为吡啶杂环氮原子的邻位或间位被c1-c3低级脂肪酸取代的取代吡啶羧酸衍生物。进一步地,其通式ⅰ为:式ⅰ。其中,r1选自-cooh、-ch2cooh或-ch2ch2cooh。可选地,含氮杂环羧酸选自:2-吡啶甲酸、3-吡啶甲酸、2-吡啶乙酸、3-吡啶乙酸、2-吡啶丙酸和3-吡啶丙酸中的任意一种。可选地,含氮杂环羧酸为2-吡啶甲酸或3-吡啶甲酸。其中,含氮杂环羧酸为吲哚杂环氮原子的邻位或间位被c1-c3低级脂肪酸取代的取代吲哚羧酸衍生物。进一步地,其通式ⅱ为:式ⅱ。其中,r2选自-cooh、-ch2cooh或-ch2ch2cooh。可选地,含氮杂环羧酸选自:2-吲哚甲酸、3-吲哚甲酸、2-吲哚乙酸、3-吲哚乙酸、2-吲哚丙酸和3-吲哚丙酸中的任意一种。可选地,含氮杂环羧酸为2-吲哚甲酸或3-吲哚甲酸。其中,含氮杂环羧酸为喹啉杂环氮原子的邻位或间位被c1-c3低级脂肪酸取代的取代喹啉羧酸衍生物。进一步地,其通式ⅲ为:式ⅲ。其中,r3选自-cooh、-ch2cooh或-ch2ch2cooh。可选地,含氮杂环羧酸选自:2-喹啉甲酸、3-喹啉甲酸、2-喹啉乙酸、3-喹啉乙酸、2-喹啉丙酸和3-喹啉丙酸中的任意一种。可选地,含氮杂环羧酸为2-喹啉甲酸或3-喹啉甲酸。可选地,邻硝基亚苄基乙酰乙酸甲酯与含氮杂环羧酸的摩尔比为1:0.02-1:0.12。例如:邻硝基亚苄基乙酰乙酸甲酯与含氮杂环羧酸的摩尔比为1:0.02、1:0.04、1:0.06、1:0.08、1:0.10或1:0.12。可选地,邻硝基亚苄基乙酰乙酸甲酯与含氮杂环羧酸的摩尔比为1:0.03-1:0.10。进一步地,邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯的摩尔比为1:0.9-1:1.3,可以使邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯反应完全,进一步减少杂质(尤其是特殊单杂)的产生。例如:邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯的摩尔比为1:0.9、1:1.1、1:1.2或1:1.3。可选地,邻硝基亚苄基乙酰乙酸甲酯与3-氨基巴豆酸甲酯的摩尔比为1:1.05-1:1.15。可选地,加成反应溶剂为c1-c4的低级脂族醇类溶剂;进一步地,反应溶剂为c1-c3的低级脂族醇类溶剂;进一步地,c1-c3的低级脂族醇类溶剂为甲醇、乙醇和异丙醇中的任何一种或两种溶剂的混合物。可选地,c1-c3的低级脂族醇类溶剂与邻硝基亚苄基乙酰乙酸甲酯的质量比为2:1-8:1,例如:c1-c3的低级脂族醇类溶剂与邻硝基亚苄基乙酰乙酸甲酯的质量比为2:1、3:1、4:1、5:1、6:1、7:1或8:1。进一步地,c1-c3的低级脂族醇类溶剂与邻硝基亚苄基乙酰乙酸甲酯的质量比为3:1-5:1。本申请中,加成反应温度为25℃至回流温度,适宜的反应温度有利于减少不应有的副反应。例如:加成反应温度为25℃、30℃、35℃、40℃、45℃、50℃、55℃或回流温度。进一步地,加成反应温度为45℃-回流温度。可选地,加成反应时间为8-14h。通过用定时取反应液作液相检测的方式来判别反应完成程度。当发现硝苯地平的峰面积不再扩大时,说明加成反应已达终点。通过上述方法,可以确定反应时间为8-14h。例如:加成反应时间为8h、9h、11h、13h或14h。可选地,加成反应时间为10-12h。保温反应结束后,冰水浴循序降温析晶,并于5-10℃陈化3h,使硝苯地平结晶能够被充分析出。例如:反应液降温至5℃、6℃、7℃、8℃、9℃或10℃。硝苯地平结晶完毕,通过分离(真空过滤或离心分离)的方式获得滤饼,然后将滤饼用预冷至10℃以下的同一醇溶剂100ml分作3次洗涤,最后进行真空干燥,得到硝苯地平。实验例一种硝苯地平的制备方法,其反应式具体如下:其中,上述反应式中,甲醇溶剂是一个具体的低级脂族醇溶剂的选择,但不仅限于甲醇溶剂,溶剂的具体选择参照表1中的溶剂条件。上述反应式中的回流10-11h均是一个具体反应时间,但不仅限于上述反应时间,因为反应条件(如催化剂)不同,反应的具体时间不同,甚至差异较大(参见表2)。进一步地,制备方法具体为:在配备带有磁力搅拌、温度计和冷凝器的容量为500ml的反应瓶中,先倒入低级脂肪醇溶剂,然后加入50.0g的邻硝基亚苄基乙酰乙酸甲酯,开启搅拌,加入含氮杂环羧酸催化剂,水浴升温至55℃后加入3-氨基巴豆酸甲酯,继续升温至回流温度后持续搅拌反应,并定时取反应液样用hplc法作跟踪检测,当目标产物面积百分比例不再增加时,视为本反应终点。其中,硝苯地平的加成反应条件如表1:表1硝苯地平的主要制备技术条件保温反应结束后,水浴降温析晶,并于5-10℃陈化3h,然后真空过滤得到滤饼,将滤饼用预冷至10℃以下的同一醇溶剂100ml分作3次(50ml+30ml+20ml)洗涤,抽干后于50-60℃真空干燥至固体含湿率≤1%后出料,得到硝苯地平粗制品。将用本发明方法得到的硝苯地平湿粗品用5-6倍量的甲醇简单重结晶一次后得到纯度和有关物质指标更佳的硝苯地平精制品(见表3、图5)。表1的加成反应时间和摩尔收率、纯度、总杂及特殊单杂的面积百分比结果列于表2:表2加成反应时间、摩尔收率、纯度、总杂及特殊单杂面积百分比结果备注:1)上表中特殊杂质(基因毒性杂质)特指起始原料邻硝基苯甲醛。从表1-表2的条件与结果中可以看出,虽然实施例提供的加成反应摩尔收率和对比例提供的摩尔收率数据差异不大,但是,实施例中的硝苯地平粗品纯度全部达到《内控质量标准》规定指标(≥98.5%),尤其是反应时间明显缩短,基因毒性杂质含量也明显降低。所以,本申请提供的制备方法可以提高硝苯地平的纯度,减少制备过程中产生的杂质,并能较大幅度地缩短反应时间。比较实施例1-实施例3的条件与结果,邻硝基亚苄基乙酰乙酸甲酯与催化剂2-吡啶甲酸的摩尔比为1:0.05时,硝苯地平的加成反应中的催化效果最佳。比较实施例1、实施例4和实施例5的条件与结果,当溶剂为甲醇时,加成反应中的催化效果最佳。比较实施例1、实施例6-实施例12的条件与结果,当催化剂为2-吡啶甲酸和2-吲哚甲酸时,催化效果优于催化剂为2-喹啉甲酸或3-喹啉甲酸的催化效果。进一步地,图1为实施例1的技术条件下的加成反应终点反应液的液相色谱图。其中,硝苯地平粗品的纯度为99.95%,特殊单杂的面积比仅为0.04%,总杂面积比也只有0.05%。说明在此条件下当以2-吡啶甲酸为加成反应催化剂时综合效果最好。图2为实施例8的技术条件下的加成反应终点反应液的液相色谱图。虽然目标物纯度和有关物质水平依然处于理想状态,不难看出,使用2-吲哚甲酸的催化效果稍逊于2-吡啶甲酸。图3为实施例11的技术条件下的加成反应终点反应液的液相色谱图。本实施例是以3-喹啉甲酸为加成反应催化剂,其效果与2-吲哚甲酸催化剂相当或稍逊。图4为对比例1的技术条件下的加成反应终点反应液的液相色谱图。结果表明,当使用经典的乙醇钠为加成反应催化剂时,即便其他反应条件相同,其目标物纯度,尤其是特殊单杂和总杂面积百分比数据与图1、图2和图3相比,图4中的各项指标数据与其差异明显,尤其是反应时间明显加长。图5为实施例8所得的硝苯地平粗品经用乙醇一次重结晶后的硝苯地平精制样品的液相色谱图。结果表明,用本发明方法获得的硝苯地平加成粗品经用乙醇作简单的一次重结晶后综合质量水平更佳。实施例1的制备方法提供的硝苯地平成品连续三批次检测结果(见)表3)。表3连续三批次硝苯地平原料药放样实验样品(精制品)检测结果备注:表3的含量数据为外标法计算结果,而非面积归一法直接测得的纯度结果。有关物质含量系按中国药典(cp2015)规定方法检测。其中,杂质d为3-氨基巴豆酸甲酯(原料杂质),系欧洲药典(ep10.0)规定的杂质检查项,cp2015尚未作规定。表3中3批样品的基因毒性杂质含量数据远远低于《标准》规定限度值,且3批样品中的杂质ⅰ、ⅱ和杂质d均未检出(含量低于检测限),其中最大单杂和总杂含量也显著低于《标准》规定限度值,足以说明本申请提供的方法具有显著的效果。表4硝苯地平的元素分析结果chn0计算值58.96%5.24%8.09%27.71%测定值58.92%5.24%8.08%27.74%从表4的内容可以看出,该批样品纯度较高。以上所描述的实施例是本申请一部分实施例,而不是全部的实施例。本申请的实施例的详细描述并非旨在限制要求保护的本申请的范围,而是仅仅表示本申请的选定实施例。基于本申请中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本申请保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1