一种制备1-O-乙酰-2,3,5-三-O-苯甲酰-1-β-D-呋喃核糖的方法与流程
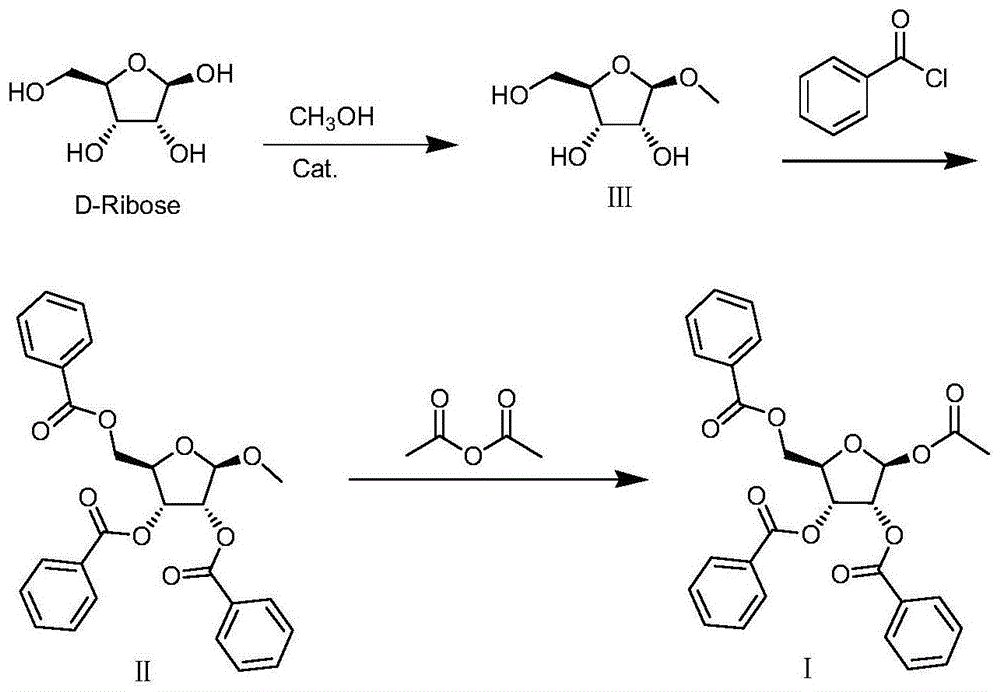
本发明涉及有机合成领域,具体为一种制备1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖的方法。
背景技术:
在化学类药物中,具有治疗疾病效果的核苷及其衍生物有着非常重要的地位,其数量呈迅速增长的趋势,其中1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖是多个医药品种的关键中间体。该产品应用广泛,例如:(1)克罗拉宾是一种新型的嘌呤核苷类抗癌药物,由美国排名前十的生物制药公司美国健赞公司(genzymecorporation,nasdaq:genz)研发成功,商品名称clofarabine,2004年12月28日美国食品与药品管理局(fda)通过快速通道批准clofarabine用于儿童顽固性或复发性急性淋巴细胞白血病的治疗,是目前唯一可以特异性用于治疗儿童急性粒细胞性白血病(all)的药物;治疗白血病有效率高,并且耐受性好,没有不可预知的不良反应。既可以静脉给药,也可以口服,为十多年来首个获准专门用于儿童的白血病治疗新药。(2)阿糖胞苷,是一种主要作用于细胞s增殖期的嘧啶类抗代谢药,对痘病毒、疱疹病毒、腺病毒等dna病毒有显著抑制作用。主要用于急性白血病,对急性粒细胞白血病疗效最好,对恶性淋巴瘤、肺癌、消化道癌、头颈部癌有一定的疗效。是目前我国临床上用于治疗急性非淋巴骨髓细胞白血病最有效的药物之一。
1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖己经在世界范围内引起了人们的广泛兴趣和高度重视,相关的研究文章也不断见诸发表。目前文献报道的制备1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖的方法大部分用吡啶作为溶剂,具有反应步骤长、环境污染大、成本高等缺点。例如常俊标等(公开号cn101177442a)从d-核糖开始用吡啶作为溶剂经四步反应合成本产品,使用了溴,红磷等危险品,还有一步需要柱分离。同时未合成得到消旋的纯品,作为中间产物往下反应。其中李俊等(申请专利号cn108570078)从腺苷出发经两步法合成,步骤少,但是原料昂贵,不适宜工业化。baud(tetrahedronletters,1990,vol.31,#31,p.4437-4440),也是用吡啶作为溶剂经三步反应合成本品,并且目前方法化学纯度普遍达不到99%。
技术实现要素:
本发明的目的在于提供一种制备1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖的方法,以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:一种制备1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖的方法,包括以下步骤:
s1:甲酯化反应:将d-核糖与甲醇及催化剂混合,加热至30-70℃反应6-24小时,检测合格后中和,浓缩得甲酯化中间体iii;
s2:苯甲酰化反应:将甲酯化中间体iii与有机溶剂及缚酸剂混合,并加入4-二甲氨基吡啶作为催化剂,冷却后滴加苯甲酰氯反应,水洗,无水硫酸钠干燥再过滤浓缩,得苯甲酰化中间体ii;
s3:乙酰化反应:将苯甲酰化中间体ii与有机溶剂以及醋酐混合,再加入催化剂,搅拌,在0℃-70℃温度下,保温反应10-20小时,反应合格后,冷却结晶,离心,得到粗品;用有机溶剂重结晶,离心烘干得所述的1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖。
优选的,所述s1)甲酯化反应中,d-核糖与催化剂量以及甲醇摩尔比为1∶(0.001-0.1)∶(10-50);其中催化剂为浓硫酸、对甲苯磺酸、甲磺酸、氢溴酸、盐酸或者hcl气体。
优选的,所述s1)甲酯化反应中,d-核糖与催化剂量以及甲醇摩尔比为1∶(0.005-0.02)∶(20-30);其中催化剂为浓硫酸或者对甲苯磺酸。
优选的,所述s2)苯甲酰化反应中,苯甲酰氯与缚酸剂及d-核糖的摩尔比为∶(3-7)∶(4-9)∶1,其中有机溶剂为三氯甲烷、1,2-二氯乙烷、二氯甲烷、乙酸乙酯、n,n-二甲基甲酰胺、三乙胺或者甲苯;其中缚酸剂为三乙胺、甲基二异丙胺、碳酸钾、碳酸钠、碳酸氢钠、或者碳酸铯。
优选的,所述s2)苯甲酰化反应中,苯甲酰氯与缚酸剂及d-核糖的摩尔比为:(3-5)∶(4-7)∶1;其中有机溶剂为甲苯、三氯甲烷或者1,2-二氯乙烷;其中缚酸剂为三乙胺和甲基二异丙胺
优选的,所述s3)乙酰化反应中,醋酸酐与催化剂及d-核糖的摩尔比为:(1-5)∶(0.001-0.5)∶1,其中催化剂为浓硫酸、对甲苯磺酸、甲磺酸或者hcl气;其中有机溶剂为乙酸、甲苯、乙酸乙酯、二氯甲烷、三氯甲烷、1,2-二氯乙烷;其中结晶溶剂为乙酸乙酯、甲醇、乙醇、异丙醇、甲苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷等。
优选的,所述s3)乙酰化反应中,醋酸酐与催化剂及d-核糖的摩尔比为:(1-2)∶(0.1-0.2)∶1;其中催化剂为浓硫酸和对甲苯磺酸;其中有机溶剂为乙酸、甲苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷;其中结晶溶剂为甲苯、乙醇、异丙醇、二氯甲烷、三氯甲烷中的一种或两种及以上混合物。
与现有技术相比,本发明的有益效果是:本发明所公布制备1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖的方法,具有反应步骤短、化学及光学纯度高、操作简单、原料易得、成本低廉、“三废”量少、对环境友好等优点,适宜工业化生产。
附图说明
图1为本发明的化学反应过程示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
请参阅图1,本发明提供一种技术方案:一种制备1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖的方法,包括以下步骤:
s1:甲酯化反应:将d-核糖与甲醇及催化剂混合,加热至30-70℃反应6-24小时,检测合格后中和,浓缩得甲酯化中间体iii;
s2:苯甲酰化反应:将甲酯化中间体iii与有机溶剂及缚酸剂混合,并加入4-二甲氨基吡啶作为催化剂,冷却后滴加苯甲酰氯反应,水洗,无水硫酸钠干燥再过滤浓缩,得苯甲酰化中间体ii;
s3:乙酰化反应:将苯甲酰化中间体ii与有机溶剂以及醋酐混合,再加入催化剂,搅拌,在0℃-70℃温度下,保温反应10-20小时,反应合格后,冷却结晶,离心,得到粗品;用有机溶剂重结晶,离心烘干得所述的1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖。
进一步的,所述s1)甲酯化反应中,d-核糖与催化剂量以及甲醇摩尔比为1∶(0.001-0.1)∶(10-50);其中催化剂为浓硫酸、对甲苯磺酸、甲磺酸、氢溴酸、盐酸或者hcl气体。
进一步的,所述s1)甲酯化反应中,d-核糖与催化剂量以及甲醇摩尔比为1∶(0.005-0.02)∶(20-30);其中催化剂为浓硫酸或者对甲苯磺酸。
进一步的,所述s2)苯甲酰化反应中,苯甲酰氯与缚酸剂及d-核糖的摩尔比为:(3-7)∶(4-9)∶1,其中有机溶剂为三氯甲烷、1,2-二氯乙烷、二氯甲烷、乙酸乙酯、n,n-二甲基甲酰胺、三乙胺或者甲苯;其中缚酸剂为三乙胺、甲基二异丙胺、碳酸钾、碳酸钠、碳酸氢钠、或者碳酸铯。
进一步的,所述s2)苯甲酰化反应中,苯甲酰氯与缚酸剂及d-核糖的摩尔比为∶(3-5)∶(4-7)∶1;其中有机溶剂为甲苯、三氯甲烷或者1,2-二氯乙烷;其中缚酸剂为三乙胺和甲基二异丙胺
进一步的,所述s3)乙酰化反应中,醋酸酐与催化剂及d-核糖的摩尔比为:(1-5)∶(0.001-0.5)∶1,其中催化剂为浓硫酸、对甲苯磺酸、甲磺酸或者hcl气;其中有机溶剂为乙酸、甲苯、乙酸乙酯、二氯甲烷、三氯甲烷、1,2-二氯乙烷;其中结晶溶剂为乙酸乙酯、甲醇、乙醇、异丙醇、甲苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷等。
进一步的,所述s3)乙酰化反应中,醋酸酐与催化剂及d-核糖的摩尔比为:(1-2)∶(0.1-0.2)∶1;其中催化剂为浓硫酸和对甲苯磺酸;其中有机溶剂为乙酸、甲苯、二氯甲烷、三氯甲烷、1,2-二氯乙烷;其中结晶溶剂为甲苯、乙醇、异丙醇、二氯甲烷、三氯甲烷中的一种或两种及以上混合物。
实施例1:
甲酯化反应:将0.09gtsoh和85.0g甲醇投入到250ml三颈瓶(搅拌,冷凝管)中,加入15.0gd-核糖,30-50℃反应合格,加入三乙胺淬灭反应,浓缩得到甲酯化中间体iii,直接用于下一步反应;
苯甲酰化反应:向装有机械搅拌器和温度计的500ml三颈瓶中,加入甲酯化中间体iii和80.0ml1,2-二氯乙烷以及72ml三乙胺,搅拌,随后加入0.15gdmap搅拌溶解;30℃滴加20ml1,2-二氯乙烷和63.0g苯甲酰氯,滴毕,30℃反应完全;后处理、过滤得到苯甲酰化中间体ii,直接用于下一步反应;
乙酰化反应:将上步得到苯甲酰化中间体ii与80ml醋酸乙酯以及25g醋酐混合,加入6.0g硫酸,50-60℃搅拌反应,合格后,过滤得粗品45.07g;重结晶烘干得白色固体30.0g1-o-乙酰-2,3,5-三-0-苯甲酰-1-β-d-呋喃核糖。
所得的1-0-乙酰-2,3,5-三-0-苯甲酰-1-β-d-呋喃核糖,其总收率59.45%,hplc纯度99.43%,熔点为129.2-130.4℃,比旋度[α]20d=+24.3°(c=1,吡啶)。
实施例2:
甲酯化反应:将0.09gtsoh和85.0g甲醇投入到250ml三颈瓶(搅拌,冷凝管)中,加入15.0gd-核糖,常温反应,加入三乙胺淬灭反应,浓缩得到甲酯化中间体iii,直接用于下一步反应;
苯甲酰化反应:向装有机械搅拌器和温度计的500ml三颈瓶中,加入甲酯化中间体iii和80.0ml二氯甲烷、75ml三乙胺,随后加入0.15gdmap搅拌溶解;40-50℃滴加20ml二氯甲烷和60.50g苯甲酰氯,滴毕,50℃反应,结束后。过滤得到苯甲酰化中间体ii,直接用于下一步反应;
乙酰化反应:将上步得到苯甲酰化中间体ii与80ml醋酸以及25.0g醋酐加入到500ml反应瓶中,加入2g硫酸,常温搅拌,反应合格后,浓缩得粗品48.0g;重结晶过滤,烘干得白色固体33.1g1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖。
所得的1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖,其总收率65.61%,hplc纯度99.55%,熔点为129.6-130.8℃,比旋度[α]20d=+24.2°(c=1,吡啶)。
实施例3:
甲酯化反应:将10g浓硫酸和8500g甲醇投入到20l反应釜(搅拌,冷凝管)中加入1500gd-核糖,加热至50℃反应,合格后加入三乙胺淬灭反应浓缩得到甲酯化中间体iii,直接用于下一步反应;
苯甲酰化反应:向装有机械搅拌器和温度计的50l反应釜中,加入甲酯化中间体iii和8l三氯甲烷、8.2l三乙胺,加入15gdmap搅拌溶解;30-40℃滴加2l三氯甲烷和6790g苯甲酰氯,滴毕,50℃反应2h,反应完全;蒸馏得到苯甲酰化中间体ii,直接用于下一步反应;
乙酰化反应:在20l反应釜中将上步得到苯甲酰化中间体ii与8l醋酸溶液混合再加入1230g醋酐,加入60g硫酸,50℃下搅拌合格后离心得粗品4732g;重结晶过滤,烘干得白色固体3122g1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖。
所得的1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖,其总收率61.85%,hplc纯度99.32%,熔点为129.1-130.3℃,比旋度[α]20d=+24.4°(c=1,吡啶)。
实施例4:
甲酯化反应:将45gtsoh和42.5kg甲醇投入到100l反应釜(搅拌,冷凝管)中,加入7.5kgd-核糖,加热至60℃反应,加入甲基二异丙胺淬灭反应浓缩得到甲酯化中间体iii,直接用于下一步反应;
苯甲酰化反应:向装有机械搅拌器和温度计的200l反应釜中,加入甲酯化中间体iii和40l甲苯、41.3l甲基二异丙胺加入75gdmap搅拌至全部溶解;40℃滴加10l甲苯和36.38kg苯甲酰氯,滴毕,50℃反应,结束后。过滤,得到苯甲酰化中间体ii,直接用于下一步反应;
乙酰化反应:在200l反应釜中将上步得到苯甲酰化中间体ii与40l乙酸混合,加入6.2kg醋酐,加入300g硫酸,50℃搅拌,检测合格后,离心得粗品23.5kg;重结晶,过滤,烘干得白色固体16.3kg1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖。
所得的1-o-乙酰-2,3,5-三-o-苯甲酰-1-β-d-呋喃核糖,其总收率64.62%,hplc纯度99.49%,熔点为129.4-130.7℃,比旋度[α]20d=+24.0°(c=1,吡啶)。
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!