一种不对称合成光学纯度的光甘草定的方法与流程
本发明涉及有机合成技术领域,具体涉及一种不对称合成光学纯度的光甘草定的方法。
背景技术:
光果甘草(glycyrrhizaglabra)系多年生草本植物,生长于欧洲南部、亚洲、地中海地带,在俄罗斯、西班牙、伊朗和印度地区广泛种植,在中国的新疆也有大面积种植。
光甘草定(glabridin)是光果甘草中的主要黄酮类成分之一。光甘草定在细胞色素p450/nadph氧化系统中显示出很强的抗自由基氧化作用,能明显抑制体内新陈代谢过程中所产生的自由基、以免对氧化敏感的生物大分子(低密度脂蛋白ldl,dna)和细胞壁等被自由基氧化损伤,从而可以防治与自由基氧化有关的某些病理变化,如动脉粥样硬化,细胞衰老等。因此光甘草定不但具有治疗过敏性炎症等抗炎和抗菌药用价值,还是目前疗效好、功能全面的化妆品原料,具有抑制黑色素原等的美白功效和抗氧化作用;此外,光甘草定还有一定的降血脂和降血压的作用。
目前,从植物提取的光甘草定大多纯度为40%;化学纯度超过97%的光甘草定呈现白色或棕黄色固体。从光果甘草中天然提取的光甘草定,英文名称为glabridin,是具有(r)-构型的光学纯度的化合物,具有如下结构式:
目前主流合成光甘草定的方法有三种:
第一条路线为韩国的sangkuyoo等人在专利wo2005/037815a1中,分别合成两个片段,然后在-78℃下发生perkin反应,和mitsunobu反应,最后在酸性条件下脱去保护基得到消旋的光甘草定。
第二条路线为日本专利jp2006008604a报道的合成路线,其主要包括了friedel-crafts酰基化反应、吡喃环成环反应、成异黄酮环、还原脱去羰基,最后在碱性条件下得到消旋的光甘草定。
第三条路线为中国的纪文华等人的专利cn103030647,其主要包括了friedel-crafts酰基化反应、吡喃环成环反应、羰基还原和最后的脱除保护基的反应。
上述三条路线的光甘草定都为消旋体,即r-和s-构型的结构各占一半,与从天然植物中分离提取的r-构型光甘草定不同。
而cn108440553公开了一种钌复合物催化的不对称合成光学纯度的光甘草定的方法,此方法以r保护基保护的异黄酮为原料,加入由金属钌、手性二胺配体、对甲基异丙基苯、三乙基硅烷、三氟醋酸等化合物合成具有(r)-构型的光学纯度的光甘草定。但此方法的原料价格昂贵,且部分如对甲基异丙基苯等原料等对人体具有毒性且容易引起燃烧、爆炸等事故。因此急需一种更为经济、安全的合成方法。
技术实现要素:
本发明针对上述现有技术存在的问题,本发明的目的是提供一种不对称合成光学纯度的光甘草定的方法。
为实现本发明的目的,通过以下技术方案予以实现:
一种不对称合成光学纯度的光甘草定的方法,包括以下步骤:
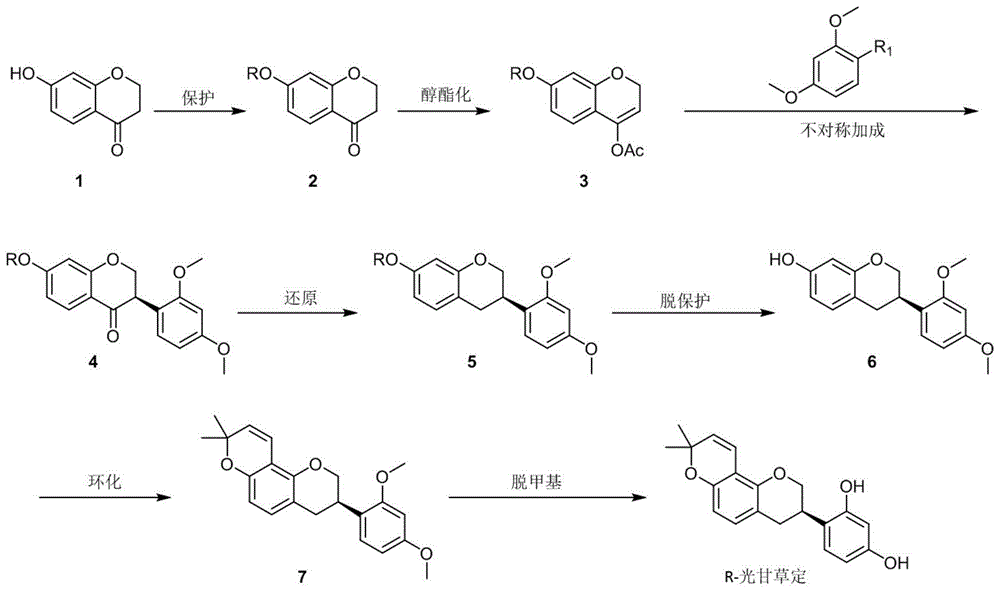
(1)7-羟基色满-4-酮和卤化苄在碱性、加热条件下反应合成,得如下式化合物2;
(2)化合物2在酸性、加热条件下进行烯醇酯化反应,得如下式化合物3;
(3)化合物3使用惰性气体保护,加入钯金属催化剂与有机磷配体反应,得下式化合物4:
所述有机磷配体如下式:
(4)化合物4使用惰性气体保护,进行羰基还原反应,得下式化合物5:
(5)化合物5进行脱除酚羟基保护基反应,得下式化合物6:
(6)化合物6进行环化反应,得下式化合物7:
(7)化合物7进行脱甲基反应,得具有光学纯度的光甘草定;
上述结构中,r代表保护基,r2代表苯基、萘基、3-氨基苯基、苄基、3-异丙基苯基、烷基及其衍生物;r3、r4分别独立代表环己基或环戊基。
优选地,在步骤(1)中,所述卤化苄为氯化苄、溴化苄、2,4-二氯氯化苄或碘化苄。
优选地,在步骤(3)中,所述有机磷配体为r-二环己基(2’-(萘-2-氧基)-[1,1’-双萘]-2-基)磷、或r-二环己基(2’-(3-氨基苯氧基)-[1,1’-双萘]-2-基)磷;所述钯金属催化剂为pd(oac)2。
进一步优选地,在步骤(1)中,在加热条件下,在有机溶剂中加入碱性物、7-羟基色满-4-酮和卤化苄反应,反应后蒸馏并纯化得化合物2。
进一步优选地,在步骤(2)中,将化合物2、乙酸丙烯酯和对甲苯磺酸置于容器中加热反应,然后冷却,蒸馏除去溶剂,将得到的油状液体溶于乙醚中,水洗,无水硫酸钠干燥,过滤,蒸馏除去溶剂,残留物用柱色谱分离纯化得到化合物3。
进一步优选地,在步骤(3)中,在惰性气体保护下,将pd(oac)2、有机磷配体、naoac、带2,4-二甲氧基苯基的有机物、化合物3加入至有机溶剂中反应,然后过滤、过层析柱分离纯化得化合物4;
所述带2,4-二甲氧基苯基的有机物为2,4-二甲氧基氯苯、2,4-二甲氧基溴苯或2,4-二甲氧基苯基三氟甲磺酸酯。
进一步优选地,在步骤(4)中,在惰性气体保护下,将化合物4与还原剂加入至有机溶剂中反应,浓缩、过滤后过层析柱分离纯化得化合物5。
更进一步优选地,所述还原剂为硼氢化钠或氢化铝锂。
进一步优选地,在所述步骤(6)中,将化合物5、碳酸钾、碘化钾和3-氯-3-甲基丁炔加入到有机溶剂中,搅拌并加热进行反应,冷却,过滤,滤液加热回流,冷却,加入乙酸乙酯稀释,洗涤,有机相用无水硫酸钠干燥,过滤,浓缩,残留物用柱色谱进行分离纯化得到化合物7。
进一步优选地,在所述步骤(7)中,向有机溶剂中加入化合物7、alcl3和nai反应,将反应液用二氯甲烷萃取,干燥,蒸馏出溶剂后得光学纯度的光甘草定。
本发明通过以7-羟基色满-4-酮为起始原料,经过烯醇酯化、用pd催化剂和有机磷配体不对称的将2,4-二甲氧基苯基引入到羰基的邻位、羰基还原、脱除酚羟基保护基和成环反应,最终得到光学纯度的光甘草定。本发明通过反应路线引入手性中心得到光学纯度的光甘草定,其反应条件温和,操作方便,适用于工业化应用。且原料易得、成本低,产物收率良好、高立体选择性。
附图说明
图1为本发明r-光甘草定的合成路线;
图2为本发明所使用的有机磷配体。
图1中的序号1、2、3、4、5、6、7分别依次代表具体实施方式中的化合物1、2、3、4、5、6、7;图2中的r2代表苯基、萘基,3-氨基苯基,苄基、3-异丙基苯基、烷基中的一种;r3、r4分别独立的代表环己基或环戊基。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。其中,附图仅用于示例性说明,表示的仅是示意图,而非实物图,不能理解为对本专利的限制。
实施例一
(1)合成7-苄氧基的色满-4-酮(化合物2)
向装有温度计和冷凝管的三口瓶中,准确称取koh固体(0.11mol)和95%的乙醇(40ml),加热至全溶,加入7-羟基色满-4-酮(0.1mol),将得到的溶液缓慢滴加到已经出现回流的氯化苄(0.125mol)的乙醇溶液中(20ml),滴加时间约1h,滴加完毕,在回流温度下继续反应1h。反应结束后,减压蒸馏除去溶剂,残留物用乙醇重结晶,得到化合物2,收率87%。
2)合成7-苄氧基-2h-色满-4-基乙酸酯(化合物3)
将化合物2(1mmol)、乙酸丙烯酯(3mmol)和对甲苯磺酸(0.1mmol)置于反应瓶中搅拌加热回流24h,冷却至室温,减压蒸馏除去溶剂,将得到的油状液体溶于乙醚中,用水洗,无水硫酸钠干燥,过滤,减压蒸除溶剂,残留物用柱色谱分离纯化得到化合物3,收率70%。
(3)化合物3合成制备化合物4:
n2保护下,将pd(oac)2(0.02mmol)、r-二环己基(2’-(萘-2-氧基)-[1,1’-双萘]-2-基)磷(0.03mmol)、naoac(0.9mmol)、2,4-二甲氧基苯基三氟甲磺酸酯(1mmol)和乙醚加入到反应瓶中,室温搅拌15mins,加入化合物3(1mmol),将反应瓶密闭,在室温下反应10h,用盛有硅藻土的漏斗过滤,减压蒸除溶剂,残留物用柱色谱进行分离纯化得到具有光学纯度的化合物4,收率88%,96%ee(对映体超量)。
(4)化合物4合成制备化合物5
n2气保护下,向200ml四氢呋喃中加入化合物4(0.01mol),搅拌,0℃下,分批加入氢氧化铝(0.1mol),加毕后,回流3h,加入乙酸乙酯猝灭反应,减压浓缩溶剂后经柱色谱分离得到化合物5,收率82%。
(5)化合物5合成制备化合物6:
向50ml的甲醇中加入化合物5(0.05mol)和10%的pd-c(0.1g),并通入h2,40-50℃反应10h后,过滤、减压蒸馏除去溶剂得到化合物6,收率94%。
(6)化合物6合成制备化合物7
将化合物5(1mmol)、碳酸钾(2.2mmol)、碘化钾(1mmol)和3-氯-3-甲基丁炔(2mmol)加入到干燥的dmf中,搅拌下加热至80℃,反应12h,冷却至室温,过滤,滤液直接进行加热回流5h。冷却降温至室温,加入乙酸乙酯稀释,用10%的稀盐酸洗涤,水洗和饱和食盐水洗。有机相用无水硫酸钠干燥,过滤,减压条件下浓缩,残留物用柱色谱进行分离纯化得到化合物7,收率86%。
(7)化合物7合成制备r-光甘草定
在0℃下,向50ml乙腈中加入化合物7(0.01mol)、alcl3(0.1mol)和nai(0.12mol),搅拌,常温反应过夜。反应结束后,将反应液加入到冰水中,用二氯甲烷萃取,无水硫酸钠干燥,减压蒸出溶剂得粗品,经过甲苯/二氯甲烷重结晶得到淡黄色固体,即r-光甘草定,收率85%,97%ee。
实施例二
化合物3、4、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物2的合成。
化合物2的合成:
向装有温度计和冷凝管的三口瓶中,准确称取0.11mol的naoh固体和40ml的95%的乙醇,加热至全溶,加入7-羟基色满-4-酮,将得到的溶液缓慢滴加到已经出现回流的0.125mol氯化苄的乙醇溶液中(20ml),滴加时间约1h,滴加完毕,在回流温度下继续反应1h。反应结束后,减压蒸馏除去溶剂,残留物用乙醇重结晶,得到化合物2,收率85%。
实施例三
化合物3、4、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物2的合成。
化合物2的合成:
室温下向100ml丙酮中加入化合物1(0.1mol)和碳酸钾(0.12mol),然后缓慢加入溴化苄(0.12mol),反应过夜。反应结束后,过滤,减压蒸除溶剂,用乙醇进行重结晶,得到化合物2,收率88%。
实施例四
化合物3、4、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物2的合成。
化合物2的合成:
向100mlch3cn中加入化合物1(0.1mol)和碳酸钾(0.12mol),加热至回流,缓慢加入2,4-二氯氯化苄(0.12mol),回流下反应3h。反应结束后,过滤,减压蒸除溶剂,用乙醇进行重结晶,得到化合物2,收率83%。
实施例五
化合物3、4、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物2的合成。
化合物2的合成:
室温下向100ml丙酮中加入化合物1(0.1mol)和碳酸钾(0.12mol),然后缓慢加入碘化苄(0.12mol),反应过夜。反应结束后,过滤,减压蒸除溶剂,用乙醇进行重结晶,得到化合物2,收率83%。
实施例六
化合物2、3、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物4的合成。
n2保护下,将pd(oac)2(0.02mmol)、r-二环己基(2’-(3-氨基苯氧基)-[1,1’-双萘]-2-基)磷(0.03mmol)、naoac(0.9mmol)、2,4-二甲氧基苯基三氟甲磺酸酯(1mmol)和乙醚加入到反应瓶中,室温搅拌15mins,加入化合物3(1mmol),将反应瓶密闭,在室温下反应10h,用盛有硅藻土的漏斗过滤,减压条件下将滤液浓缩,残留物用硅胶柱进行分离纯化得到具有光学纯度的化合物4,收率84%,98%ee。
实施例七
化合物2、3、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物4的合成。
n2保护下,将pd(oac)2(0.02mmol)、r-二环己基(2’-(萘-2-氧基)-[1,1’-双萘]-2-基)磷(0.03mmol)、naoac(0.9mmol)、2,4-二甲氧基苯基三氟甲磺酸酯(1mmol)和乙醚加入到反应瓶中,室温搅拌15mins,加入化合物3(1mmol),将反应瓶密闭,在室温下反应10h,用盛有硅藻土的漏斗过滤,减压条件下将滤液浓缩,残留物用硅胶柱进行分离纯化得到具有光学纯度的化合物4,收率90%,92%ee。
实施例八
化合物2、3、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物4的合成。
n2保护下,将pd(oac)2(0.02mmol)、r-二环己基(2’-(萘-2-氧基)-[1,1’-双萘]-2-基)磷(0.03mmol)、naoac(0.9mmol)、2,4-二甲氧基溴苯(1mmol)和乙醚加入到反应瓶中,室温搅拌15mins,加入化合物3(1mmol),将反应瓶密闭,在室温下反应10h,用盛有硅藻土的漏斗过滤,减压条件下将滤液浓缩,残留物用硅胶柱进行分离纯化得到具有光学纯度的化合物4,收率90%,86%ee。
实施例九
化合物2、3、5、6、7和r-光甘草定的合成同实施例1,区别在于化合物4的合成。
n2保护下,将pd(oac)2(0.02mmol)、r-二环己基(2’-(萘-2-氧基)-[1,1’-双萘]-2-基)磷(0.03mmol)、naoac(0.9mmol)、2,4-二甲氧基氯苯(1mmol)和乙醚加入到反应瓶中,室温搅拌15mins,加入化合物3(1mmol),将反应瓶密闭,在室温下反应10h,用盛有硅藻土的漏斗过滤,减压条件下将滤液浓缩,残留物用硅胶柱进行分离纯化得到具有光学纯度的化合物4,收率90%,85%ee。
实施例十
化合物2、3、4、6、7和r-光甘草定的合成同实施例1,区别在于化合物5的合成。
n2气保护下,向四氢呋喃(200ml)中加入化合物4(0.01mol),搅拌,室温下,加入硼氢化钠(0.1mol)和alcl3(0.1mol),加毕后,加热回流3h,加入稀盐酸猝灭反应,减压浓缩溶剂后经柱色谱分离得到化合物5,收率76%。
实施例十一
化合物2、3、4、6、7和r-光甘草定的合成同实施例1,区别在于化合物5的合成。
n2气保护,0℃下将硼氢化钠(0.5mol)分批加入到三氟乙酸中(50ml),然后将得到的混合物滴加到化合物4(0.1mol)的二氯甲烷(40ml)溶液中,加毕后,室温反应24h,反应液用氯化钠(2m)猝灭,二氯甲烷萃取,无水硫酸钠干燥,过滤,减压蒸除溶剂,残留物用柱色谱分离纯化得到化合物5,收率67%。
实施例十二
化合物2、3、4、5、7和r-光甘草定的合成同实施例1,区别在于化合物6的合成。
n2气保护,室温下向二氯甲烷(100ml)中,加入化合物5(0.1mol)和无水fecl3(10.0mol),反应10h,过滤,减压蒸馏除去溶剂,残留物经过柱色谱纯化得到化合物6,收率90%。
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!