酰胺类化合物及其制备方法和应用与流程
1.本发明涉及药物制备技术领域,特别涉及酰胺类化合物及其制备方法和应用。
背景技术:
2.干扰素是机体受病毒或其他干扰素诱导刺激巨噬细胞、淋巴细胞及体细胞产生的具有高活性的多功能糖蛋白。干扰素作用于动物机体细胞内的干扰素受体,经信号传导等一系列的生物化学过程,启动基因合成抗病毒蛋白,抑制病毒多肽链的合成,阻断了病毒的繁殖,使病毒不能在动物机体内生长与繁殖,从而起到抗病毒的作用。干扰素对所作用的对象具有一定的选择性,产生干扰素的动物细胞只对同种或同类属动物细胞有抑制病毒作用。
3.目前干扰素主要使用外源性干扰素,但是外源性干扰素具有一些缺点,比如生产较困难,产量低,价格昂贵以及生物活性也比较低。此外,外源性干扰素有一定的毒性,可造成动物白细胞减少、肝功能异常、损害神经系统等不利影响,给畜牧生产造成严重损失。而使用干扰素诱导剂诱导动物体内产生内源性干扰素,则可以克服使用外源性干扰素的不利作用。开发相应的干扰素诱导剂具有巨大需求。
技术实现要素:
4.基于此,有必要提供一种酰胺类化合物及其制备方法和应用。该酰胺类化合物能够诱导动物体内内源性干扰素的产生,具有开发为具有抗病毒功效的产品的潜能。
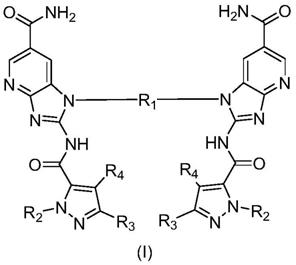
5.一种酰胺类化合物,具有式(i)所示结构:
[0006][0007]
其中,r1为c
2~10
烷烃或c
2~10
烯烃,所述c
2~10
烷烃和c
2~10
烯烃任选进一步被以下基团取代:c
1-6
烷基、c
1-6
烷氧基、含有3-8个环原子的环烷基、羰基、羟基、三氟甲基或卤素;
[0008]
r2、r3和r4各自独立地为h或c
1-6
烷基。
[0009]
上述酰胺类化合物的制备方法,包括以下步骤:
[0010][0011]
提供式(i-1)所示的化合物;
[0012]
将式(i-1)所示的化合物和h2n-r
1-nh2进行缩合反应,制得式(i-2)所示的化合物;
[0013]
将式(i-2)所示的化合物进行硝基还原反应,制得式(i-3)所示的化合物;
[0014]
使式(i-3)所示的化合物和式(i-4)所示的化合物进行缩合反应,制得式(i)所示的化合物。
[0015]
上述酰胺类化合物在制备干扰素诱导剂中的应用。
[0016]
在其中一实施例中,所述干扰素诱导剂为抗病毒药物。
[0017]
在其中一实施例中,所述干扰素诱导剂为抗肿瘤药物。
[0018]
上述酰胺类化合物在制备抗病毒药物或饲料中的应用。
[0019]
一种干扰素诱导剂,包括上述酰胺类化合物或其药学上可接受的盐。
[0020]
一种药物,包括上述酰胺类化合物或其药学上可接受的盐,与药学上可接受的辅料。
[0021]
一种饲料,包括上述酰胺类化合物。
[0022]
一种治疗方法,包括施加有效量的上述酰胺类化合物。
[0023]
上述酰胺类化合物能够有效地诱导动物体内产生干扰素,可以克服使用外源性干扰素的不利作用。
具体实施方式
[0024]
为了便于理解本发明,下面将对本发明进行更全面的描述,并给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
[0025]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0026]
定义和通用术语
[0027]
除非有相反陈述,否则下列用在说明书和权利要求书中的术语具有下述含义。
[0028]
本发明中所述的“取代”表示被一个或多个基团所替代。当多个基团从同一系列候选取代基中选择时,它们可以相同,也可以不同。
[0029]
本发明中所述的“任选地”表示所定义基团可从一系列候选基团中进行选择,也可以不选。
[0030]“烷基”是指饱和脂肪族烃基,包括直链和支链基团。c
1-c6烷基是指含有1至6个碳原子的烷基。非限定性实施例包括:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、正戊基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙基、2-甲基丁基、3-甲基丁基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2-乙基丁基、2-甲基戊基、3-甲基戊基、4-甲基戊基、2,3-二甲基丁基。c
1-c4烷基是指含有1至4个碳原子的烷基。在一实施例中,c
1-c4烷基为甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基。烷基可以是取代的或未取代的,当被取代时,取代基可以在任何可使用的连接点上被取代。
[0031]
本发明的化合物可以以非溶剂化形式和含有药学上可接受的溶剂(如水、乙醇等)的溶剂化形式存在,即包括溶剂化和非溶剂化形式。
[0032]
本发明中,某可取代位点可被一个或多个取代基取代,且当该可取代位点存在多个取代基时,多个取代基可以彼此相同或不同。
[0033]“药物组合物”表示含有一种或多种本文所述化合物或其生理学上/可药用的盐或前体药物与其他化学组分的混合物,以及其他组分。例如生理学/可药用的载体和赋形剂。药物组合物的目的是促进对生物体的给药,利于活性成分的吸收进而发挥生物活性。
[0034]
组合物中所含有的赋形剂,可以为一种或多种缓冲剂、稳定剂、抗粘剂、表面活性剂、润湿剂、润滑剂、乳化剂、粘合剂、悬浮剂、崩解剂、填充剂、吸附剂、涂料(肠的或缓释的)防腐剂、抗氧化剂,不透明的剂、助流剂、加工助剂、着色剂、甜味剂、芳香剂、调味剂和其它已知的添加剂。
[0035]“药学上可接受的盐”即“可药用的盐”,是指医药上可接受的化合物的有机或无机盐。
[0036]
当化合物是酸性或包括足够酸性生物电子等排体时,适当的“可药用的盐”指从医药上可接受的包括无机碱和有机碱的无毒碱中制备的盐。该盐衍生自含有铝、铵、钙、铜、铁、铁、锂、镁、锰盐、锰、钾、钠、辛等的无机碱。特定的实施方式包括铵、钙、镁、钾和钠盐。盐衍生自医药上可接受的有机无毒碱,该有机无毒碱包括一级、二级和三级胺的盐、包括自然存在的取代胺的取代胺、环胺和碱性离子交换树脂,如精氨酸、甜菜碱、咖啡因、胆碱、n,n.sup.1-二苄基乙二胺、乙二胺、2-二乙氨基乙醇、2-二甲基氨基乙醇、乙醇胺、乙二胺、n-乙基吗啉、n-乙基六氢吡啶、还原葡糖胺、氨基葡萄糖、组氨酸、海巴明、异丙胺、赖氨酸、葡甲胺、吗啉、哌嗪、哌啶、聚胺树脂、普鲁卡因、嘌呤、可可碱、三乙基胺、三甲胺、三丙胺、氨丁三醇等等。
[0037]
当化合物是碱性的或包括足够碱性生物电子等排体时,盐可以从医药上可接受的无毒酸中制备,包括无机和有机酸。这样的酸包括乙酸、苯磺酸、苯甲酸、樟脑磺酸、柠檬酸、乙磺酸、富马酸、葡萄糖酸、氢溴酸、盐酸、羟乙磺酸、乳酸、马来酸、苹果酸、苦杏仁酸,甲基磺酸、粘液酸、硝酸、双羟萘酸、泛酸、磷酸、硫酸、琥珀酸、酒石酸、对甲苯磺酸等等。特定的实施方式包括柠檬酸、氢溴酸、盐酸、磷酸、硫酸、马来酸、酒石酸。其它示例性的盐包括但不
限于硫酸盐、柠檬酸盐、乙酸盐、草酸盐、氯化物、溴化物、碘化物、硝酸盐,硫酸盐,磷酸盐、酸性磷酸盐、异烟酸、乳酸、水杨酸盐、酸性柠檬酸盐、酒石酸盐、油酸盐、鞣酸盐、泛酸盐、酒石酸氢盐、抗坏血酸盐、琥珀酸盐、富马酸盐、马来酸盐、龙胆酸盐、葡萄糖酸盐,葡萄糖醛酸盐、糖酸盐、甲酸盐、苯甲酸盐、谷氨酸盐、甲基磺酸盐、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐和双羟萘酸盐(例如,1,1'-亚甲基-双-(2-羟基-3-萘甲酸盐))。
[0038]
另外,包含化合物的药物制剂可以为片剂、胶囊剂、口服液体剂、丸剂、颗粒剂、散剂、软膏剂、贴剂、栓剂、口含片、滴眼剂、眼膏剂、眼膏剂、滴耳剂、喷剂、气雾剂、吸入剂、注射剂等。
[0039]
术语“治疗有效量”是指有效化合物或药物试剂的用量,改善、治愈或治疗疾病或病症的一种或多种症状的必要的最小量。
[0040]
另外,本发明所述的化合物和药物组合物可单独给药,也可以与其他药剂联合施用。对于与一种以上的活性剂的联合治疗,当该活性剂在分开的剂量制剂中时,该活性剂可以分开施用或联合施用。另外,一种药剂的施用可在另一种药剂施用之前、同时或之后进行。当与其他药剂联合施用时,第二药剂的“有效量”将视所用药物的类型而定。
[0041]
施用途径
[0042]
本发明的一种或多种化合物通过适合于受治疗的生物体(如猫)的任何途径施用。合适的途径包括口服、直肠、鼻、肺、局部(包括口腔和舌下)、和胃肠外(包括皮下、肌内)等。
[0043]
本发明的化合物或药物组合物也可以包含在试剂盒中。
[0044]
详细说明
[0045]
本发明提供了一种酰胺类化合物,具有式(i)所示结构:
[0046][0047]
其中,r1为c
2~10
烷烃或c
2~10
烯烃,所述c
2~10
烷烃和c
2~10
烯烃任选进一步被以下基团取代:c
1-6
烷基、c
1-6
烷氧基、含有3-8个环原子的环烷基、羰基、氰基、羟基、三氟甲基、或卤素;
[0048]
r2、r3和r4各自独立地为h或c
1-6
烷基。
[0049]
进一步地,r1为c
2~10
的烷烃或c
2~10
烯烃,c
2~10
的烷烃或c
2~10
烯烃任选进一步被以下基团取代:c
1-4
烷基、c
1-4
烷氧基、卤素、羟基、羰基或氰基。
[0050]
更进一步地,r1为c
3~5
的烷烃或c
3~5
烯烃。
[0051]
更进一步地,r1为丁基或c
1-3
烷基取代丁基;更进一步地,r1为丁基。
[0052]
进一步地,r2、r3和r4中至少有两个为c
1-6
烷基。
[0053]
更进一步地,r3和r4中一个为h,一个为c
1-6
烷基。
[0054]
进一步地,r2和r3各自独立地为c
1-4
烷基;更进一步地,r2为乙基,r3为甲基。
[0055]
更进一步地,上述酰胺类化合物具有式(ii)所示结构:
[0056][0057]
本发明还提供了上述酰胺类化合物的制备方法,包括以下步骤:
[0058]
s101:提供式(i-1)所示的化合物;
[0059][0060]
其中,式(i-1)所示的化合物可以采用市售原料或采用现有的方法合成。
[0061]
优选采用以下方法制备:
[0062]
s1011:使5-硝基-6-羟基-烟酸和二氯亚砜进行取代反应,制得5-硝基-6-氯-烟酸;
[0063][0064]
进一步地,优选步骤s1011的溶剂为dmf,二氯亚砜的添加量为10eq-20eq,更优选为14eq-16eq。
[0065]
进一步地,优选步骤s1011的操作为:将5-硝基-6-羟基-烟酸溶解在溶剂中,冰浴下缓慢滴加二氯亚砜,滴加完毕后,升温至75℃-85℃(优选80℃),反应完全后,后处理即得5-硝基-6-氯-烟酸。
[0066]
s1012:使5-硝基-6-氯-烟酸的羧酸进行酰胺化反应,制得5-硝基-6-氯-烟酰胺。
[0067][0068]
可理解的,步骤s1012中的酰胺反应可以采用本领域的常规反应,例如羧酸与胺进行缩合,在此不进行特别限定,应理解为均在本发明的保护范围内。优选步骤s1012的操作为:
[0069]
将5-硝基-6-氯-烟酸溶于有机溶剂中,缓慢滴加二氯亚砜,滴加完毕后,升温至75℃-85℃(优选80℃),反应完全后,后处理即得5-硝基-6-氯-烟酸。优选二氯亚砜的添加量为15eq-25eq(即5-硝基-6-氯-烟酸和二氯亚砜的摩尔比为1:15-25);更优选为18eq-22eq。
[0070]
通过先采用二氯亚砜进行取代反应,再采用二氯亚砜活化羧基,以利于与胺缩合,生成酰胺。如此,经取代反应后的产物可以直接用于后续酰胺化反应,无需进行额外纯化,降低操作难度,提高产率。
[0071]
另外,需要说明的是,步骤s1011中可以对产物进行后处理,也可以不对产物进行后处理,直接进行步骤s1012,应理解为均在本发明的保护范围内。
[0072]
s102:将式(i-1)所示的化合物和h2n-r
1-nh2进行缩合反应,制得式(i-2)所示的化合物;
[0073][0074]
步骤s102中缩合反应试剂可以采用常规试剂,在此不进行具体限定,优选采用diea。另外,优选式(i-1)所示的化合物和h2n-r
1-nh2的摩尔比为1:(0.3-0.6)。
[0075]
s103:将式(i-2)所示的化合物进行硝基还原反应,制得式(i-3)所示的化合物;
[0076][0077]
进一步地,优选步骤s103中采用pd/c还原;更进一步地,步骤s103包括以下步骤:
[0078]
将式(i-2)所示的化合物、甲酸铵、pd/c和溶剂混合,在温度为35℃-45℃的条件下进行反应,反应完全后,纯化分离即可。其中,优选溶剂为醇类溶剂和n-甲基吡咯烷酮(nmp)的混合液,优选醇类溶剂和nmp的体积比为1:(0.5-0.7);更优选为1:0.6;更优选醇类溶剂为甲醇。
[0079]
s104:使式(i-3)所示的化合物和式(i-4)所示的化合物进行缩合反应,制得式(i)所示的化合物。
[0080][0081]
进一步地,步骤s104包括以下步骤:将式(i-3)所示的化合物溶于有机溶剂中,加入式(i-4)所示的化合物,在冰浴下搅拌20min-40min,加入缩合剂和碱,移除冰浴,反应,至
反应完全,分离提纯即可。
[0082]
步骤s104中缩合反应的缩合剂无特别限定,优选为(1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐)(edc),缩合反应中的碱优选为三乙胺。更进一步地,式(i-3)所示的化合物和式(i-4)所示的化合物的摩尔比优选为1:(2-2.2);式(i-3)所示的化合物与缩合剂的摩尔比优选为1:(2-2.2);式(i-3)所示的化合物与碱的摩尔比优选为1:(2.8-3.2)。
[0083]
本发明还提供了上述酰胺类化合物在制备干扰素诱导剂中的应用。
[0084]
本发明还提供了一种干扰素诱导剂,包括上述酰胺类化合物。
[0085]
酰胺类化合物及其制备方法如上所述,在此不再进行赘述。
[0086]
本发明还提供了上述酰胺类化合物在制备抗病毒药物或饲料中的应用。
[0087]
进一步地,抗病毒药物为抗猪病毒性疾病的药物。更进一步地,猪病毒性疾病为猪流行性腹泻、传染性胃肠炎、轮状病毒感染疾病、猪瘟、蓝耳病、圆环病毒疾病、伪狂犬病、猪流感、水疱病或细小病毒病。
[0088]
可理解的,上述药物还可以包括药学上可接受辅料,药物可以根据需要制成有相应的剂型。
[0089]
本发明还提供了上述酰胺类化合物在制备抗病毒饲料中的应用。
[0090]
进一步地,抗病毒饲料为抗猪病毒性疾病的饲料。更进一步地,猪病毒性疾病为猪流行性腹泻、传染性胃肠炎、轮状病毒感染疾病、猪瘟、蓝耳病、圆环病毒疾病、伪狂犬病、猪流感、水疱病或细小病毒病。
[0091]
本发明还提供了一种治疗疾病的方法,包括施加有效量的上述酰胺类化合物的步骤。
[0092]
下面列举具体实施例来对本发明进行说明。
[0093]
实施例1:干扰素诱导剂(ii)的制备
[0094]
5-硝基-6-氯-烟酸的合成
[0095][0096]
在0~10℃下,往化合物5-硝基-6-羟基-烟酸(20g,108.63mmol,1eq)和socl2(196.80g,1.65mol,120.00ml,15.23eq)的混合物中滴加dmf(30ml),加完后将反应加热到80℃搅拌1h。lcms和tlc(乙酸乙酯:甲醇=10:1)监测反应完全后,旋蒸除去大部分的socl2,再小心倒入冰水(200ml)中淬灭,用乙酸乙酯(100ml*2)萃取后旋干得到化合物5-硝基-6-氯-烟酸(20g)。ms:203(m+1)
[0097]
5-硝基-6-氯-烟酰胺的合成
[0098][0099]
将化合物5-硝基-6-氯-烟酸(18g,88.87mmol,1eq)和socl2(147.60g,1.24mol,90.00ml,13.96eq)的混合物加热至80℃搅拌1小时。旋蒸除去大部分socl2,剩余物用thf
(90ml)稀释后降温至-78~-70℃,在此温度下将nh3.h2o(81.90g,654.27mmol,90.00ml,28%purity,7.36eq)小心滴加到上述体系中,滴加过程伴有大量固体析出,加完后,继续搅拌0.5小时后回温至20~30℃。将固体过滤出来,滤饼用水洗涤后干燥收集得到化合物5-硝基-6-氯-烟酰胺(14g,69.46mmol,78.16%yield),ms:202(m+1)。
[0100]
中间体a的合成
[0101][0102]
将化合物5-硝基-6-氯-烟酰胺(14.5g,71.94mmol,1eq),23a和diea(18.59g,143.87mmol,25.06ml,2eq)溶于dmf(140ml)中,将体系温度升至80℃搅拌1小时。lcms监测反应完全后,将反应液小心倒入500ml水中并伴随有大量固体析出。将固体滤出得到化合物a(13g,31.07mmol,43.20%yield),ms:419(m+1)。
[0103]
中间体(b)的合成
[0104][0105]
将化合物a(13g,31.07mmol,1eq),ammonia;formic acid(19.59g,310.73mmol,10eq)和pd/c(1.3g,31.07mmol,10%purity,1eq)溶于meoh(150ml)和nmp(70ml)中,将体系加热至40~50℃搅拌24小时。lcms监测反应完全后,先过滤除去钯碳,再旋蒸除去甲醇,将剩余物小心倒入500ml水中并伴随有大量固体析出。将固体滤出得到化合物b(10g,27.90mmol,89.79%yield)。ms:359(m+1),1h nmr(500mhz,dmso-d6)δ7.96(d,2h),7.53(s,2h),7.09(d,2h),6.86(s,2h),6.04~6.06(m,2h),4.79(s,4h),3.29~3.32(m,4h),1.65(m,4h).
[0106]
化合物(ii)的合成
[0107][0108]
将化合物b(10g,27.90mmol,1eq)和化合物c(11.44g,58.59mmol,2.1eq)溶于dmf(100ml)中,在20~30℃下搅拌0.5h。在0~5℃下,往反应体系中依次加入edci(13.37g,69.75mmol,2.5eq)和dipea(18.03g,139.51mmol,24.30ml,5eq),加完后反应体系在20~30℃下搅拌2小时。lcms监测反应完全后,将体系小心倒入500ml水中并伴有大量固体析出。将
固体过滤出来,收集后用200ml乙腈打浆后得到化合物ii(8.1g,11.58mmol,41.51%yield,97.33%purity)。1h nmr(500mhz,dmso-d6)δ12.83(s,2h),8.70~8.74(d,j=1.7hz,2h),8.09~8.12(m,4h),7.51(s,2h),6.61(s,2h),4.55(q,j=7.0hz,4h),4.25(s,4h),2.10(s,6h),1.92(s,4h),1.30(t,j=7.1hz,6h).
[0109]
实施例2:化合物(ii)诱导仔猪产生干扰素的实验研究
[0110]
材料及方法:
[0111]
选取总共24只4周龄的一次性断奶仔猪,作为试验仔猪,随机分为3组,每组8只,分别设为x组、y组、z组。其中x组为阴性对照组,受试物为生理盐水;y组受试物为干扰素诱导剂实验组,注射0.3mg/kg剂量的化合物(ii);z组为阳性药组,受试物为聚胞苷,注射0.7mg/kg剂量的聚胞苷溶液。连续注射2天后,采集仔猪血液样品测试血液中的干扰素α(ifn-α)水平。
[0112]
统计学方法:
[0113]
采用spss11.5统计软体进行统计分析,实验数据采用t检验,以p<0.05为差异有统计学意义。
[0114]
检测方法:
[0115]
采用静脉采血方式分别采集x组、y组和z组试验仔猪的血液样品,干扰素检测试剂采用ifn-α试剂盒(abcam公司的interferon beta pig elisa kit)。检测仪器为全自动多功能酶标仪(multiskan mk3,thermo,usa),测定结果如下表1所示。
[0116]
表1各试验组血清中ifn-α检测结果:
[0117]
组别受试物例数(只)血清ifn-α(ng/ml)px生理盐水81.07
±
0.14-y化合物ii84.26
±
0.53<0.05z聚胞苷82.43
±
0.31<0.05
[0118]
由表1可以看到,与阴性对照组相比,本发明提供的化合物ii可以对仔猪体内干扰素α的分泌起到显著的诱导作用,从而增强仔猪抵抗病毒感染能力,并且显著的高于聚胞苷的诱导作用。
[0119]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0120]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1