一种米拉贝隆杂质化合物及其制备方法和用途与流程
本发明属于化学药物合成技术领域,具体涉及一种米拉贝隆杂质化合物及其制备方法和用途。
背景技术:
米拉贝隆,化学名(r)-2-(2-氨基噻唑-4-基)-n-(4-{2-[(2-羟基-2-苯基乙基)氨基]乙基}苯基)乙酰胺,分子式c21h24n4o2s,分子量396.51,cas登记号223673-61-8,是日本山之内制药株式会社开发的口服有效的新型选择性β3肾上腺素能受体激动剂,于2011年首次在日本上市,2012年6月经fda批准用于治疗成年人膀胱过度活动症(oab),其结构如下所示:
专利cn1711085a报道米拉贝隆的合成方法为:以r-扁桃酸和对硝基苯乙胺为起始原料,经酰胺缩合制得式8化合物,然后经硼烷还原制得式9化合物,再经催化氢化还原制得如式7化合物,最后与2-氨基噻唑-4-乙酸缩合制得米拉贝隆,具体合成路线如下所示:
该工艺会产生含有一种二聚的工艺杂质,其性质与米拉贝隆十分相近,重结晶等常规纯化方法难以将其除尽,该杂质的存在对米拉贝隆的质量影响很大,故有效地控制和除去该二聚杂质是米拉贝隆质量控制的关键。
技术实现要素:
为了克服现有技术存在的缺陷,本发明提供了一种米拉贝隆杂质化合物或其盐、其制备方法和用途。
发明人经大量的实验发现,米拉贝隆的质量控制在分析方法的建立上必须使用二聚工艺杂质对照品进行定位,因而二聚工艺杂质是米拉贝隆质量控制的必需品;此外,杂质的存在还有可能引起严重的副反应,因而本领域迫切需要有效鉴定米拉贝隆合成中产生的杂质。
本发明最终通过以下技术方案解决上述技术问题的。
本发明提供了一种米拉贝隆杂质式i化合物或其盐:
优选的,所述盐为米拉贝隆杂质式i化合物与酸形成的盐,所述酸为无机酸或有机酸;所述无机酸优选盐酸、氢溴酸、氢碘酸、硫酸、硝酸或磷酸;所述有机酸优选甲酸、乙酸、丙酸、草酸、琥珀酸、苹果酸、富马酸、乳酸、柠檬酸、酒石酸、苦味酸、甲磺酸、乙磺酸、对甲苯磺酸等。
本发明还提供了一种米拉贝隆杂质式i化合物或其盐的制备方法,包括以下步骤:溶剂中,在缩合剂作用下,将式6化合物或其活性酯与式7化合物或其盐进行缩合反应得到,其反应方程式如下:
优选的,所述的溶剂为水和/或有机溶剂;更优选的,当式7化合物以盐酸盐的形式进行反应时,所述溶剂为水;更优选的,所述有机溶剂优选酰胺类溶剂、酮类溶剂和亚砜类溶剂中的一种或多种;进一步优选的,所述酰胺类溶剂为n,n-二甲基甲酰胺和/n,n-二甲基乙酰胺;
优选的,所述的缩合剂为二环己基碳二亚胺(dcc)、二异丙基碳二亚胺(dic)、1-(3-二甲胺基丙基)-3-乙基碳二亚胺盐酸盐(edci·hcl)、碳基二咪唑(cdi)、二苯基磷酰叠氮(dppa)和二乙基磷酰氰化物(depc)中的一种或多种;
优选的,所述的缩合剂与式6化合物或其活性酯的摩尔比较优的为1:1~2:1;
优选的,所述的缩合剂与式7化合物或其盐酸盐的摩尔比较优的为1:1~2:1;
优选的,所述的溶剂与式7化合物或其盐酸盐的体积质量比为5ml/g~50ml/g,优选10ml/g~20ml/g;
优选的,所述的反应温度为20~30℃;
优选的,式6的活性酯为式6化合物与1-羟基苯并三唑或与1-羟基-7-偶氮苯并三氮唑形成的酯;
优选的,所述方法包括如下步骤:将式6化合物、式7化合物、缩合剂和溶剂混合,加入酸调ph至2~4,10~40℃搅拌反应,加入碱调ph至8~11,搅拌,过滤,干燥;优选的,进一步还包括精制步骤,如用乙醇精制;所述缩合剂优选edci·hcl;所述溶剂优选水;所述酸优选浓盐酸;所述碱优选氢氧化钠;
其反应进程可以采用本领域中常规的检测方法(如tlc,hplc或nmr)进行监控,一般以如式7化合物或其盐酸盐消失时为反应终点,所述反应时间较优的为0.5~3小时。
进一步的,本发明还提供了式6化合物的制备方法,包括如下步骤:有机溶剂中,在酸试剂的作用下,将式5化合物进行酸解脱boc反应得到,其反应方程式如下:
优选的,所述的有机溶剂为醚类溶剂、芳香烃类溶剂、卤代烃类溶剂(如二氯甲烷)、酰胺类溶剂、酯类溶剂、亚砜类溶剂和醇类溶剂中的一种或多种;
更优选的,所述醚类溶剂为四氢呋喃、乙醚、乙二醇二甲醚、二噁烷和2-甲基四氢呋喃中的一种或多种;
更优选的,所述的芳香烃类溶剂为苯、甲苯和二甲苯中的一种或多种。所述的卤代烃类溶剂为二氯甲烷、三氯甲烷和1,2-二氯乙烷中的一种或多种;
更优选的,所述的酰胺类溶剂为n,n-二甲基甲酰胺和/或n,n-二甲基乙酰胺;
更优选的,所述的酯类溶剂选自乙酸乙酯、乙酸异丙酯和乙酸异戊酯中的一种或多种;
更优选的,所述的亚砜类溶剂为二甲基亚砜;
更优选的,所述的醇类溶剂为甲醇、乙醇或异丙醇;
优选的,所述酸试剂可选本领域常规的无机酸或有机酸一种或多种;
更优选的,所述的无机酸为盐酸、氢溴酸、氢碘酸、硫酸、硝酸或磷酸中的一种或多种;
更优选的,所述的有机酸为甲酸、乙酸、丙酸、草酸、丙二酸、柠檬酸、酒石酸、碳酸、甲磺酸、乙磺酸或三氟乙酸中的一种或多种。
优选的,所述的溶剂与式5化合物体积质量比为3ml/g~20ml/g,优选范围3ml/g~8ml/g;
优选的,所述酸试剂与式5化合物的体积质量比为2~5ml/g;
优选的,所述酸解脱boc反应的温度为10~40℃,优选范围20~30℃;
优选的,所述式6化合物的制备方法包括如下步骤:将式5化合物溶于有机溶剂,加入酸试剂,10~40℃下搅拌反应,去有机溶剂,加入碱溶液,室温搅拌,过滤,滤液用酸调ph至4-6,析出固体,过滤干燥;所述碱优选饱和碳酸钠;所述酸优选盐酸(如1mol/l盐酸);所述酸试剂优选三氟乙酸;所述去有机溶剂优选减压抽除方式。
其反应进程可以采用本领域中常规的检测方法(如tlc,hplc或nmr)进行监控,一般以如式5化合物消失时为反应终点,所述反应时间较优为1~4小时。
进一步的,本发明还提供了式5化合物的制备方法,包括如下步骤:在溶剂中,在碱试剂的作用下,将式4化合物进行酯水解反应得到,其反应方程式如下:
优选的,所述的溶剂为水和/或有机溶剂;
更优选的,所述的有机溶剂为醚类溶剂、芳香烃类溶剂、卤代烃类溶剂、酰胺类溶剂、酯类溶剂、亚砜类溶剂和醇类溶剂中的一种或多种;
进一步优选的,所述的醚类溶剂为四氢呋喃、乙醚、乙二醇二甲醚、二噁烷和2-甲基四氢呋喃中的一种或多种;
进一步优选的,所述的芳香烃类溶剂为苯、甲苯和二甲苯中的一种或多种;
进一步优选的,所述的卤代烃类溶剂为二氯甲烷、三氯甲烷和1,2-二氯乙烷中的一种或多种;
进一步优选的,所述的酰胺类溶剂为n,n-二甲基甲酰胺和/或n,n-二甲基乙酰胺;
进一步优选的,所述的酯类溶剂选自乙酸乙酯、乙酸异丙酯和乙酸异戊酯中的一种或多种;进一步优选的,所述的亚砜类溶剂为二甲基亚砜;
进一步优选的,所述的醇类溶剂为甲醇、乙醇或异丙醇;
更优选的,所述溶剂为水和乙醇;
优选的,所述碱试剂可选本领域常规的无机碱或有机碱的一种或多种;
更优选的,所述的无机碱为碳酸钠、碳酸氢钠、碳酸氢钾、碳酸钾、氢氧化钾、lioh或其水合物、氢氧化钠和氢氧化锂中的一种或多种;进一步优选的,所述无机碱为lioh或其水合物;
更优选的,所述的有机碱为甲醇钠、乙醇钠、叔丁醇钾,叔丁醇钠、二异丙基乙胺,三乙胺,吡啶,4-二甲基吡啶(dmap)中的一种或多种。
优选的,所述的溶剂与式4化合物体积质量比为2ml/g~5ml/g;
优选的,所述碱试剂与式4化合物的摩尔比为2:1~4:1;
优选的,所述水解反应的温度为30℃~50℃;
优选的,所述式5化合物的制备方法包括如下步骤:将式4化合物、无机碱和有机溶剂混合,在30-50℃下搅拌反应1~6h;优选的,还包括后处理步骤,后处理步骤包括但不限于:将反应液用水稀释,酸调ph至5~6,搅拌,过滤,洗涤,干燥;所述无机碱优选lioh一水合物;所述酸优选冰醋酸;调ph步骤优选在冰浴下进行;所述酸优选冰醋酸;所述洗涤优选采用水。
其反应进程可以采用本领域中常规的检测方法(如tlc,hplc或nmr)进行监控,一般以如式4化合物消失时为反应终点,所述反应时间较优为3~6小时。
进一步的,本发明还提供了式4化合物的制备方法,包括如下步骤:溶剂中,在缩合剂的作用下,将式2化合物与式3化合物或其活性酯进行缩合反应得到,其反应方程式如下:
优选的,所述的溶剂为水和/或有机溶剂,所述的有机溶剂优选为醚类溶剂、酰胺类溶剂、酮类溶剂和亚砜类溶剂中的一种或多种;
更优选的,所述的醚类溶剂为四氢呋喃、乙醚、乙二醇二甲醚、二噁烷和2-甲基四氢呋喃中的一种或多种;
更优选的,所述的酰胺类溶剂较优的为n,n-二甲基甲酰胺(dmf)和/n,n-二甲基乙酰胺;
更优选的,所述的酮类溶剂为n-甲基吡咯烷酮;
更优选的,所述的亚砜类溶剂较优的为二甲基亚砜;
优选的,所述的缩合剂为二环己基碳二亚胺、二异丙基碳二亚胺、1-(3-二甲胺基丙基)-3-乙基碳二亚胺盐酸盐(edci·hcl)、羰基二咪唑、二苯基磷酰叠氮和二乙基磷酰氰化物中的一种或多种;更优选的,所述的缩合剂为1-(3-二甲胺基丙基)-3-乙基碳二亚胺盐酸盐(edci·hcl);
优选的,所述的缩合剂与式3化合物的摩尔比为1:1~1:2;
优选的,所述的溶剂与式3化合物的体积质量比为2~20ml/g,优选范围3~8ml/g;
优选的,所述缩合反应的温度为40℃~60℃;
优选的,式3化合物的活性酯为式3化合物与1-羟基苯并三唑或1-羟基-7-偶氮苯并三唑形成的酯;
优选的,所述式4化合物的制备方法包括如下步骤:将式3化合物、式2化合物、缩合剂(如edci·hcl)和有机溶剂混合,40~60℃搅拌反应;优选的,还包括后处理步骤,后处理步骤包括但不限于稀释、萃取、洗涤、干燥、抽除溶剂,更优选的,稀释采用水;萃取采用乙酸乙酯;洗涤采用盐酸和饱和食盐水;抽除溶剂在减压条件下进行;有机溶剂优选酰胺类溶剂(如dmf)。
其反应进程可以采用本领域中常规的检测方法(如tlc,hplc或nmr)进行监控,一般以如式3化合物或其盐酸盐消失时为反应终点,所述反应时间较优的为24~28小时。
进一步的,本发明还提供了式3化合物的制备方法,包括如下步骤:溶剂中,在碱的作用下,将式2化合物与boc酸酐(二碳酸二叔丁酯)进行反应得到,其反应方程式如下:
优选的,所述的溶剂为水和/或有机溶剂;所述的有机溶剂为醚类溶剂、芳香烃类溶剂、卤代烃类溶剂、酰胺类溶剂、酯类溶剂、亚砜类溶剂和醇类溶剂中的一种或多种;
更优选的,所述的醚类溶剂为四氢呋喃、乙醚、乙二醇二甲醚、二噁烷和2-甲基四氢呋喃中的一种或多种;
更优选的,所述的芳香烃类溶剂为苯、甲苯和二甲苯中的一种或多种;
更优选的,所述的卤代烃类溶剂为二氯甲烷、三氯甲烷和1,2-二氯乙烷中的一种或多种;
更优选的,所述的酰胺类溶剂为n,n-二甲基甲酰胺和/或n,n-二甲基乙酰胺;
更优选的,所述的酯类溶剂选自乙酸乙酯、乙酸异丙酯和乙酸异戊酯中的一种或多种;
更优选的,所述的亚砜类溶剂为二甲基亚砜;
更优选的,所述的醇类溶剂为甲醇、乙醇或异丙醇。
优选的,所述碱可选本领域常规的无机碱和/或有机碱的一种或多种;
更优选的,所述的无机碱为碳酸钠,碳酸氢钠,碳酸氢钾,碳酸钾,氢氧化钾,氢氧化钠和氢氧化锂中的一种或多种;
更优选的,所述的有机碱为甲醇钠、乙醇钠、叔丁醇钾,叔丁醇钠、二异丙基乙胺,三乙胺,吡啶,4-二甲基吡啶(dmap)中的一种或多种。
优选的,所述的boc酸酐与式2化合物的摩尔比为1:1~2:1;
优选的,所述的碱与式2化合物的摩尔比为1:1~3:1;
优选的,所述的溶剂体积与式2化合物质量比为3~20ml/g,优选范围5~8ml/g;
优选的,所述缩合反应的温度为10℃~40℃,更优选为20~30℃;
优选的,式3化合物的制备方法包括如下步骤:将式2化合物、碱溶于有机溶剂,加入boc酸酐(二碳酸二叔丁酯),10~40℃搅拌反应;优选的,还包括后处理步骤,后处理步骤包括但不限于:洗涤、干燥、抽除溶剂、调ph值(如至5~6)、搅拌。
其反应进程可以采用本领域中常规的检测方法(如tlc,hplc或nmr)进行监控,一般以如式2化合物消失时为反应终点,所述反应时间较优的为3~10小时。
本发明还提供式i化合物的制备方法,包括由式2化合物制备式3化合物、由式3化合物制备式4化合物、由式4化合物制备式5化合物、由式5化合物制备式6化合物、由式6化合物制备式7化合物、由式7化合物制备式i化合物的步骤,其反应方程式如下:
优选的,由式2化合物制备式3化合物、由式3化合物制备式4化合物、由式4化合物制备式5化合物、由式5化合物制备式6化合物、由式6化合物制备式7化合物、由式7化合物制备式i化合物的步骤同上所述。
另一方面,本发明还提供式ⅰ化合物的用途,该化合物用作米拉贝隆质量控制的杂质化合物的对照品或用于米拉贝隆的杂质鉴定。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:
1.本发明提供了全新的米拉贝隆杂质化合物,纯度较高,可用作米拉贝隆质量控制的杂质化合物对照品,能够有效鉴定米拉贝隆合成中产生的杂质,并对杂质化合物进行定量控制。
2.本发明制备方法,反应条件温和,操作简单,该合成方法的副反应少,产品收率和纯度较高,所以可以快速低成本的获取高质量的产品。
附图说明
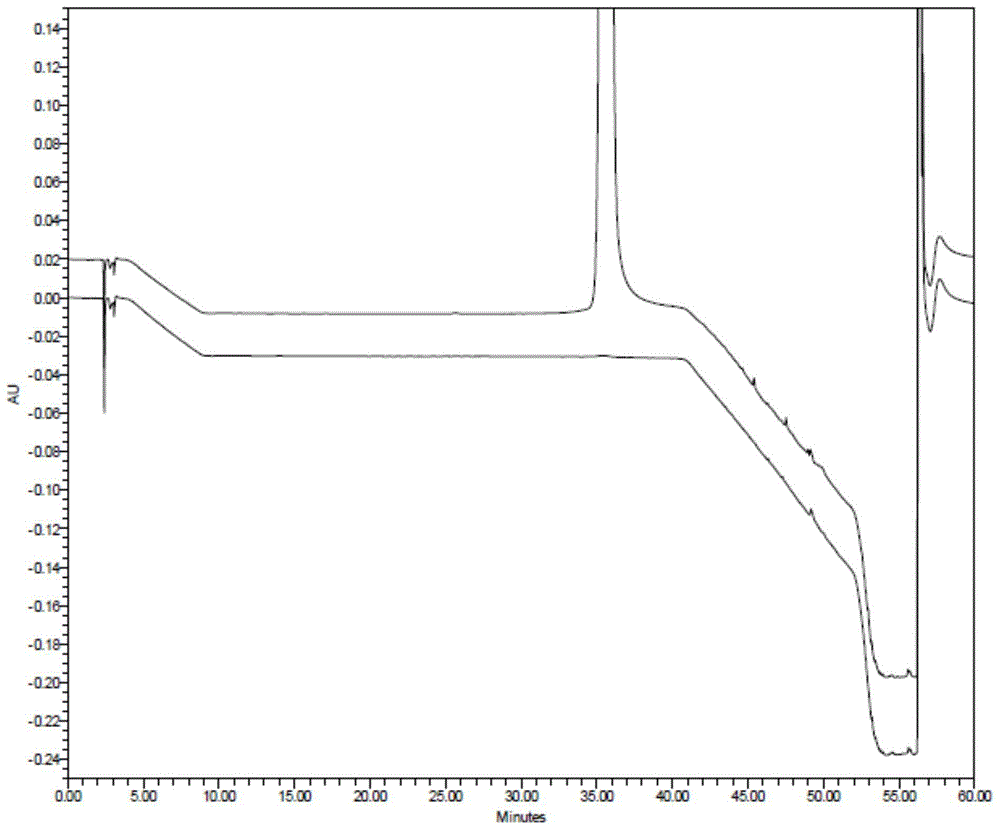
图1所表示的为实例14制得的米拉贝隆精制品与空白对照的hplc谱图
图2所表示的为实例9制得的式i化合物、实例14制得的米拉贝隆粗品以及米拉贝隆精制品的hplc谱图
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
下列实施例中1h-nmr谱使用bruker-400核磁共振仪,内标是四甲基硅烷,化学位移δ(ppm)表示;质谱用agilent6210液相色谱-飞行时间质谱联用仪。
实施例1-2式3化合物的制备
实施例1
将化合物2(20.00g,107mmol)和4-二甲氨基吡啶(16.00g,131mmol)溶于二氯甲烷100ml,20℃搅拌下滴加二碳酸二叔丁酯(28.20g,129mmol),滴完20℃搅拌反应.反应完毕后依次用100ml1mol/l盐酸、饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压抽除溶剂,所得残余物和lioh一水合物(9.00g,214mmol)、乙醇50ml及水25ml混合,40℃下搅拌2h。反应完毕后,加60ml水稀释,冰浴下滴加冰醋酸调节ph至5~6,浓缩除去乙醇,加入200ml水,继续搅拌1h后过滤,40ml水洗,干燥,得到19.87g化合物3,白色固体,收率80.5%。
实施例2
将化合物2(40.00g,214mmol)和4-二甲氨基吡啶(32.00g,262mmol)溶于二氯甲烷320ml,30℃搅拌下滴加二碳酸二叔丁酯(56.60g,261mmol),滴完30℃搅拌反应.反应完毕后依次用200ml1mol/l盐酸、饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压抽除溶剂,所得残余物和lioh一水合物(18.00g,428mmol)、乙醇100ml及水50ml混合,40℃下搅拌2h。反应完毕后,加120ml水稀释,冰浴下滴加冰醋酸调节ph至5~6,浓缩除去乙醇,加入400ml水,继续搅拌1h后过滤,80ml水洗,干燥,得到40.26g化合物3,白色固体,收率81.6%。
实施例3-4式4化合物的制备
实施例3
将化合物3(13.87g,54.8mmol),化合物2(2-氨基噻唑-4-乙酸乙酯,11.10g,59.6mmol)和edci·hcl(9.45g,54.8mmol)溶于42mldmf中,于40℃加热搅拌24h.反应完毕后加80ml水稀释,乙酸乙酯萃取(3×50ml),有机相依次用50ml1mol/l盐酸、饱和食盐水洗涤,无水硫酸钠干燥,减压抽除溶剂,得到17.56g化合物4,黄色固体,收率75.1%。
实施例4
将化合物3(27.74g,110mmol),化合物2(2-氨基噻唑-4-乙酸乙酯,22.20g,120mmol)和edci·hcl(37.80g,220mmol)溶于220mldmf中,于60℃加热搅拌24h.反应完毕后加160ml水稀释,乙酸乙酯萃取(3×100ml),有机相依次用100ml1mol/l盐酸、饱和食盐水洗涤,无水硫酸钠干燥,减压抽除溶剂,得到35.62g化合物4,黄色固体,收率76.2%。
实施例5-6式5化合物的制备
实施例5
向250ml反应烧瓶中加入化合物4(14.63g,34.3mmol),再加入lioh一水合物(3.75g,89.4mmol)、乙醇35ml和水20ml,于30℃下加热搅拌3h。反应完毕后加30ml水稀释,冰浴下滴加冰醋酸调节ph至5~6,继续搅拌1h后过滤,50ml水洗,干燥,得到12.23g化合物5,淡黄色固体,收率89.5%。
实施例6
向500ml反应烧瓶中加入化合物4(29.26g,68.6mmol),再加入lioh一水合物(11.51g,274mmol)、乙醇80ml和水50ml,于50℃下加热搅拌3h。反应完毕后加60ml水稀释,冰浴下滴加冰醋酸调节ph至5~6,继续搅拌1h后过滤,100ml水洗,干燥,得到24.21g化合物5,淡黄色固体,收率88.5%。
实施例7-8式6化合物的制备
实施例7
向250ml反应烧瓶中加入化合物5(12.23g,30.6mmol)与50ml二氯甲烷,加入37ml三氟乙酸,20℃下搅拌2h后减压抽除溶剂,加入50ml饱和碳酸钠溶液,室温搅拌0.5h,过滤,滤液用1mol/l盐酸调节ph至5,析出固体,过滤,少量水洗滤饼,干燥得8.68g化合物6,淡黄色固体,收率95.1%,esi-ms(m/z):299.02[m+h]+;1hnmr(dmso-d6)δ:12.361(s,1h),12.183(s,1h),6.951(s,2h),6.928(s,1h),6.329(m,1h),3.588(s,2h),3.563(s,2h)。
实施例8
向250ml反应烧瓶中加入化合物5(6.12g,15.3mmol)与49ml二氯甲烷,加入30.6ml三氟乙酸,30℃下搅拌2h后减压抽除溶剂,加入30ml饱和碳酸钠溶液,室温搅拌0.5h,过滤,滤液用1mol/l盐酸调节ph至5,析出固体,过滤,少量水洗滤饼,干燥得4.30g化合物6,淡黄色固体,收率94.2%。
实施例9-10式ⅰ化合物的制备
实施例9
向250ml反应烧瓶中加入化合物6(2.00g,6.70mmol)、化合物7(2.00g,6.83mmol)、edci·hcl(1.44g,7.51mmol)和20ml水,用5ml浓盐酸调节溶液ph至3,室温20℃搅拌3h,滴加2mol/l氢氧化钠溶液调节ph至9,继续搅0.5h后过滤,少量水洗,干燥,粗品用乙醇精制得到1.42g化合物ⅰ,类白色固体,收率62.1%。纯度98.70%;esi-ms(m/z):537.25[m+h]+,269.22[(m+2h)/2]+;1hnmr(dmso-d6)δ:10.078(s,1h),7.520(s,1h),7.499(s,1h),7.285-7.344(m,4h),7.202-7.245(m,1h),6.932-6.942(m,3h),6.321(s,1h),4.622-4.654(m,1h),3.676(s,2h),3.563(s,2h),2.727-2.831(m,2h),2.644-2.708(m,4h)。
实施例10
向250ml反应烧瓶中加入化合物6(4.00g,13.4mmol)、化合物7(4.00g,13.7mmol)、edci·hcl(2.87g,15.0mmol)和80ml水,用7ml浓盐酸调节溶液ph至3,室温30℃搅拌3h,滴加2mol/l氢氧化钠溶液调节ph至9,继续搅0.5h后过滤,少量水洗,干燥,粗品用乙醇精制得到1.42g化合物ⅰ,类白色固体,收率97.31%,纯度98.73%。
实施例11-13式ⅰ化合物相关盐的制备
实施例11
向100ml反应烧瓶中加入化合物i(2.00g,3.73mmol)和20ml乙醇,搅拌下溶清,滴加1.0ml浓盐酸,析出类白色固体,过滤,少量乙醇洗,干燥,得到2.03g化合物ⅰ的盐酸盐,类白色固体,收率95.01%。纯度99.20%。
实施例12
向100ml反应烧瓶中加入化合物i(2.00g,3.73mmol)和20ml乙醇,搅拌下溶清,滴加2.0ml48%氢溴酸,析出类白色固体,过滤,少量乙醇洗,干燥,得到2.12g化合物ⅰ的氢溴酸盐,类白色固体,收率92.35%。纯度99.31%。
实施例13
向100ml反应烧瓶中加入化合物i(2.00g,3.73mmol)和20ml乙醇,搅拌下溶清,滴加1.79g甲磺酸,析出类白色固体,过滤,少量乙醇洗,干燥,得到2.24g化合物ⅰ的盐酸盐,类白色固体,收率94.58%。纯度99.26%。
实施例14米拉贝隆的制备与精制
在室温下,在8.00g的r-2-[[2-(4氨基苯基)]氨基]-1-苯基乙醇盐酸盐、4.32g的2-氨基噻唑-4-乙酸、2.64g的浓盐酸及120ml的水混合液中加入5.76g的1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐(edci.hcl),搅拌1小时。然后在反应液中滴加2.40g氢氧化钠及40ml水的混合液,进行析晶,滤取生成的固体,即为米拉贝隆粗品,用水洗涤后直接进行精制(纯度99.71%)。
将上述湿品用90ml乙醇和140ml水混合液即热至80℃溶解,降温至50℃加入5.0%晶种,然后降温至20℃,过滤固体,50℃真空干燥得到白色固体米拉贝隆精制品(纯度99.91%)。
取实施例14中制得的米拉贝隆粗品、实施例9中制得的式i化合物,采用高效液相色谱法进行检测,结果见表1。
表1中,序号1表示为实施例14中制得的米拉贝隆粗品的保留时间、峰面积、相对峰面积、峰高等信息;序号2表示为实施例9中制得的式i化合物的保留时间、峰面积、相对峰面积、峰高等信息;序号3和4表示为实施例14中制得的米拉贝隆粗品中其它未知杂质a、b的保留时间、峰面积、相对峰面积、峰高等信息。
表1式i化合物与米拉贝隆粗品及其它杂质相关信息表
取实施例14中制得的米拉贝隆精制品、实施例9中制得的式i化合物,采用高效液相色谱法进行检测,结果见表2。
表2中,序号1表示为实施例14中制得的米拉贝隆精制品的保留时间、峰面积、相对峰面积、峰高等信息;序号2表示为实施例9中制得的式i化合物化合物的保留时间、峰面积、相对峰面积、峰高等信息;序号3和4表示为实施例14中制得的米拉贝隆精制品中其他杂质的保留时间、峰面积、相对峰面积、峰高等信息。
表2式i化合物与米拉贝隆精制品及其它杂质相关信息表
实施例14制得的米拉贝隆精制品中hplc纯度大于99.91%,杂质(如式ⅰ所示的化合物)含量小于0.05%,满足药品质量的要求。
取实例14制得的米拉贝隆精制品与空白对照品进行hplc检测,结果见图1;
取实例9制得的式i化合物、实例14制得的米拉贝隆粗品以及米拉贝隆精制品进行hplc检测,结果见图2;
由表1、表2、图1、图2的数据看出,本发明的米拉贝隆有关物质能够有效的鉴定米拉贝隆合成中产生的杂质,从而控制米拉贝隆的药品质量。
- 还没有人留言评论。精彩留言会获得点赞!