消旋多取代二氢异喹啉的不对称氢化动力学拆分方法
1.本发明涉及不对称催化氢化领域,具体涉及消旋多取代二氢异喹啉的不对称氢化动力学拆分方法。
背景技术:
2.光学纯的四氢异喹啉类骨架结构广泛存在于天然产物和药物中(j.d.scott,r.m.williams,chem.rev.2002,102,1669;m.chrzanowska,m.d.rozwadowska,chem.rev.2004,104,3341;m.chrzanowska,a.grajewska,m.d.rozwadowska,chem.rev.2016,116,12369),其不对称合成受到科学家的广泛关注。二氢异喹啉(c.li,j.xiao,j.am.chem.soc.2008,130,13208;m.chang,w.li,x.zhang,angew.chem.int.ed.2011,50,10679)或者异喹啉(s.-m.lu,y.-q.wang,x.-w.han,y.-g.zhou,angew.chem.int.ed.2006,45,2260;l.shi,z.-s.ye,l.-l.cao,r.-n.guo,y.hu,y.-g.zhou,angew.chem.int.ed.2012,51,8286;z.-s.ye,r.-n.guo,x.-f.cai,m.-w.chen,l.shi,y.-g.zhou,angew.chem.int.ed.2013,52,3685)的直接不对称氢化已发展成为制备手性四氢异喹啉最高效的方法,但这些方法仅限于合成单取代或双取代的手性四氢异喹啉化合物。对于手性三取代四氢异喹啉的催化不对称合成仍然缺乏有效的方法。目前仅有几例通过手性源试剂合成的报道,但这些方法通常合成步骤繁冗,产率较低,还有很大的底物局限性(d.br
ó
zda,koroniak,m.d.rozwadowska,tetrahedron:asymmetry 2000,11,3017;m.nicoletti,d.o’hagan,a.m.z.slawin,j.chem.soc.perkin trans.1,2002,116;t.saitoh,k.shikiya,y.horiguchi,t.sano,chem.pharm.bull.2003,51,667;t.ramanivas,g.gayatri,d.priyanka,v.l.nayak,k.k.singarapu,a.k.srivastava,rsc adv.2015,5,73373;s.g.davies,a.m.fletcher,a.b.frost,m.s.kennedy,p.m.roberts,j.e.thomson,tetrahedron 2016,72,2139;v.erdmann,b.r.lichman,j.zhao,r.c.simon,w.kroutil,j.m.ward,h.c.hailes,d.rother,angew.chem.int.ed.2017,56,12503)。
3.因此,发展一种不使用昂贵的手性起始原料或手性拆分试剂、操作简单易行、产物收率高、产物光学纯度高的新型手性多取代四氢异喹啉的高效不对称合成方法是本领域亟待解决的问题。
技术实现要素:
4.本发明的目的在于提供一种高效的消旋多取代二氢异喹啉类化合物的不对称氢化动力学拆分方法,从而为高对映选择性地合成多取代手性四氢异喹啉类化合物和相应的多取代手性二氢异喹啉类化合物提供新方法。
5.为了实现上述目的,本发明第一方面提供一种含多取代二氢异喹啉类化合物对映异构体的混合物的不对称氢化动力学拆分方法,该方法包括:在手性二胺金属催化剂存在下,采用氢气对含多取代二氢异喹啉类化合物对映异构体的混合物进行不对称氢化处理,得到多取代手性四氢异喹啉类化合物和多取代手性二氢异喹啉类化合物的单一光学异构
体;其中,所述多取代二氢异喹啉类化合物为式(1)所示的化合物;所述多取代手性四氢异喹啉类化合物为式(2)所示的化合物;
6.式(1)式(2)
7.其中,r1、r2和r3各自独立地为取代的或未取代的c
1-c
10
的烷基、取代的或未取代的c
3-c
10
的环烷基、取代的或者未取代的芳基或者取代的或未取代的芳苄基且r3可为氢,r4、r5和r6各自独立地为氢、卤素、取代的或未取代的c
1-c
10
的烷基、取代的或未取代的c
3-c
10
的环烷基、取代的或者未取代的芳基或者取代的或未取代的芳苄基,或者r5与r4或r6环合成环,其中,对于取代的烷基、取代的环烷基、取代的芳基和取代的芳苄基中的取代基各自独立地选自卤素、硝基、羟基、c1-c6的烷基、c1-c6的羟烷基、c1-c6的烷氧基、c1-c6的卤代烷基和c2-c6的酰胺基中的一种或多种。
8.本发明第二方面提供了一种多取代手性四氢异喹啉类化合物,所述多取代手性四氢异喹啉类化合物为式(2)所示的化合物;
9.式(2)各个基团如上文所描述的。
10.本发明第三方面提供了一种多取代手性二氢异喹啉类化合物的单一光学异构体,其特征在于,所述多取代手性二氢异喹啉类化合物为式(1)所示的化合物;
11.式(1)各个基团如上文所描述的。
12.本发明的方法,通过采用手性二胺金属催化剂催化不对称加氢的方式,实现含多取代二氢异喹啉类化合物对映异构体的混合物,特别是外消旋的含多取代二氢异喹啉类化合物的混合物实现拆分,可同时获得具有一定光学纯度的多取代手性四氢异喹啉类化合物和多取代手性二氢异喹啉类化合物的单一光学异构体;且本发明具有操作简单易行,条件温和,高效,高对映选择性,拆分系数高等优点。
具体实施方式
13.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各
个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
14.本发明提供一种含多取代二氢异喹啉类化合物对映异构体的混合物的不对称氢化动力学拆分方法,该方法包括:在手性二胺金属催化剂存在下,采用氢气对含多取代二氢异喹啉类化合物对映异构体的混合物进行不对称氢化处理,得到多取代手性四氢异喹啉类化合物和多取代手性二氢异喹啉类化合物的单一光学异构体;其中,所述多取代二氢异喹啉类化合物为式(1)所示的化合物;所述多取代手性四氢异喹啉类化合物为式(2)所示的化合物;
15.式(1)式(2)
16.其中,r1、r2和r3各自独立地为取代的或未取代的c
1-c
10
的烷基、取代的或未取代的c
3-c
10
的环烷基、取代的或者未取代的芳基或者取代的或未取代的芳苄基且r3可为氢,r4、r5和r6各自独立地为氢、卤素、取代的或未取代的c
1-c
10
的烷基、取代的或未取代的c
3-c
10
的环烷基、取代的或者未取代的芳基或者取代的或未取代的芳苄基,或者r5与r4或r6环合成环,其中,对于取代的烷基、取代的环烷基、取代的芳基和取代的芳苄基中的取代基各自独立地选自卤素、硝基、羟基、c1-c6的烷基、c1-c6的羟烷基、c1-c6的烷氧基、c1-c6的卤代烷基和c2-c6的酰胺基中的一种或多种。
17.根据本发明,优选情况下,r1、r2和r3各自独立地为取代的或未取代的c
1-c6的烷基、取代的或未取代的c
3-c6的环烷基、取代的或者未取代的芳基或者取代的或未取代的芳苄基且r3可为氢,r4、r5和r6各自独立地为氢、卤素、取代的或未取代的c
1-c6的烷基、取代的或未取代的c
3-c6的环烷基、取代的或者未取代的芳基或者取代的或未取代的芳苄基,或者r5与r4或r6环合成3-8元环;其中,对于取代的烷基、取代的环烷基、取代的芳基和取代的芳苄基中的取代基各自独立地选自卤素、硝基、羟基、c1-c4的烷基、c1-c4的羟烷基、c1-c4的烷氧基、c1-c4的卤代烷基和c2-c4的酰胺基中的一种或多种;
18.更优选地,r1、r2和r3各自独立地为甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、取代的或者未取代的苯基或取代的或者未取代的苄基且r3可为氢,r4、r5和r6各自独立地为氢、氟、氯、溴、碘、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、取代的或者未取代的苯基或取代的或者未取代的苄基,或者r5与r4或r6环合成6-8元芳环;其中,对于取代的苯基和取代的苄基中的取代基各自独立地选自氟、氯、溴、碘、硝基、羟基、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、-cf3、-ccl3、-cbr3、-ch2cf3、-ch2ccl3、-ch2cbr3、-nh-co-ch3和-nh-co-ch2ch3中的一种或多种。
19.其中,r5可与r4或r6环合成苯环结构。
20.根据本发明,本发明的加氢底物是含多取代二氢异喹啉类化合物对映异构体的混
合物,该混合物中具有至少一对对映异构体,特别是该混合物为外消旋体,而本发明的方法可以从这样的混合物中拆分获得单一光学异构体,并同时制得多取代手性四氢异喹啉类化合物。
21.其中,式(1)所示化合物中的3和4位可以是手心碳位点,该手心碳位点可以是r-构型,也可以是s-构型,含多取代二氢异喹啉类化合物对映异构体的混合物则可以是3r,4r-光学构型与3s,4s-光学构型的混合物,也可以是3r,4s-光学构型与3s,4r-光学构型的混合物,但并不排除包括少量其他构型;也可以是4种构型的混合物。
22.式(1)
23.优选情况下,式(1)和(2)中,r2和r3为顺式取代。
24.在本发明的一种优选的实施方式中,所述多取代二氢异喹啉类化合物选自下式所示化合物:
25.[0026][0027]
在本发明另一种优选的实施方式中,所述多取代二氢异喹啉类化合物的单一光学异构体选自下式所示化合物:
[0028]
[0029][0030]
优选情况下,所述多取代手性四氢异喹啉类化合物中,r1、r2和r3为顺式取代。
[0031]
在本发明的一种优选实施方式中,所述多取代手性四氢异喹啉类化合物选自下式所示化合物:
[0032][0033][0034]
根据本发明,含多取代二氢异喹啉类化合物对映异构体的混合物可以是市售品,也可以是本领域常规方法制得的多取代二氢异喹啉类化合物对映异构体的外消旋体,本发
明对此并无特别的限定,例如可以采用以下方程式所示的两种合成路线中的一种制备。
[0035]
(1)
[0036]
(2)
[0037]
所述手性二胺金属催化剂优选采用cn105111208a中公开的手性催化剂,其中,所述手性二胺金属催化剂优选为式(4)所示结构的化合物中的至少一种:
[0038]
式(4)
[0039]
其中,在式(4)中,
[0040]
m选自金属钌、铑或铱;
[0041]
l1选自取代或未取代的η
6-苯配位基、取代或未取代的η
5-茂配位基;
[0042]
且l1中任选存在的取代基各自独立地选自c1-c10的烷基中的至少一种;
[0043]
x选自cl-、br-、i-、ch3coo-、no
3-、hso
4-、h2po
4-、bf
4-、sbf
6-、pf
6-、二(三氟甲磺酰)亚胺负离子、三氟甲磺酸负离子、取代或未取代的c24-c32四芳基硼负离子、取代或未取代的c12-c36二芳基磷酸负离子、取代或未取代的c12-c36联芳基二酚衍生的磷酸负离子;且x中任选存在的取代基各自独立地选自氟、氯、溴、硝基、甲基、甲氧基、三氟甲基、羟基和乙酰氨基中的至少一种。
[0044]
根据本发明,所述催化剂是如式(4)所示结构的钌(ru)、铑(rh)或铱(ir)的配合物,作为式(4)所示结构的配合物的配位基之一的式(5)是由二胺nhr
”-
连接臂-nhso2r'形成,其中,-nhso2r'一端的n与m形成共价键,而nhr
”-
一端的n与m形成配位键,从而形成了式(4)所示的配位基。其中,在手性催化剂中,所述二胺nhr
”-
连接臂-nhso2r'中的连接臂可以使得该连接臂上与nhr
”-
端的n连接的碳原子和/或-nhso2r'端的n连接的碳原子成为手性中心,从而使得作为手性催化剂的式(4)所示结构的化合物具有一定的催化选择性,特别是(r,r)-构型、(s,s)-构型、(r)-构型或(s)-构型的这类化合物适用于催化式(1)所示结构的化合物。例如采用(r,r)或(r)-手性二胺nhr
”-
连接臂-nhso2r'作为手性催化剂时,通常将提高(s,s)-式(5)所示结构的化合物的对映体过量值,而当采用(s,s)或(s)-手性二胺nhr
”-
连接臂-nhso2r'作为手性催化剂时,通常将提高(r,r)-式(5)所示结构
的化合物的对映体过量值。不过,通常一对对映体的催化剂的催化效果相反,例如如果(r,r)-型催化剂的催化效果是提高(s,s)型的产物含量,那么(s,s)-型的催化剂的催化效果则是提高(r,r)型的产物含量,对此本领域技术人员应当理解。
[0045]
为了更有利于配合本发明的式(1)所示结构的消旋多取代二氢异喹啉类化合物的催化氢化,优选情况下,定义式(4)中的配位基为式(5)所示的基团,
[0046]
式(5)
[0047]
形成式(5)所示的配位基的化合物选自以下化合物中的至少一种:
[0048]
式(i-1)式(i-2)式(i-3)
[0049]
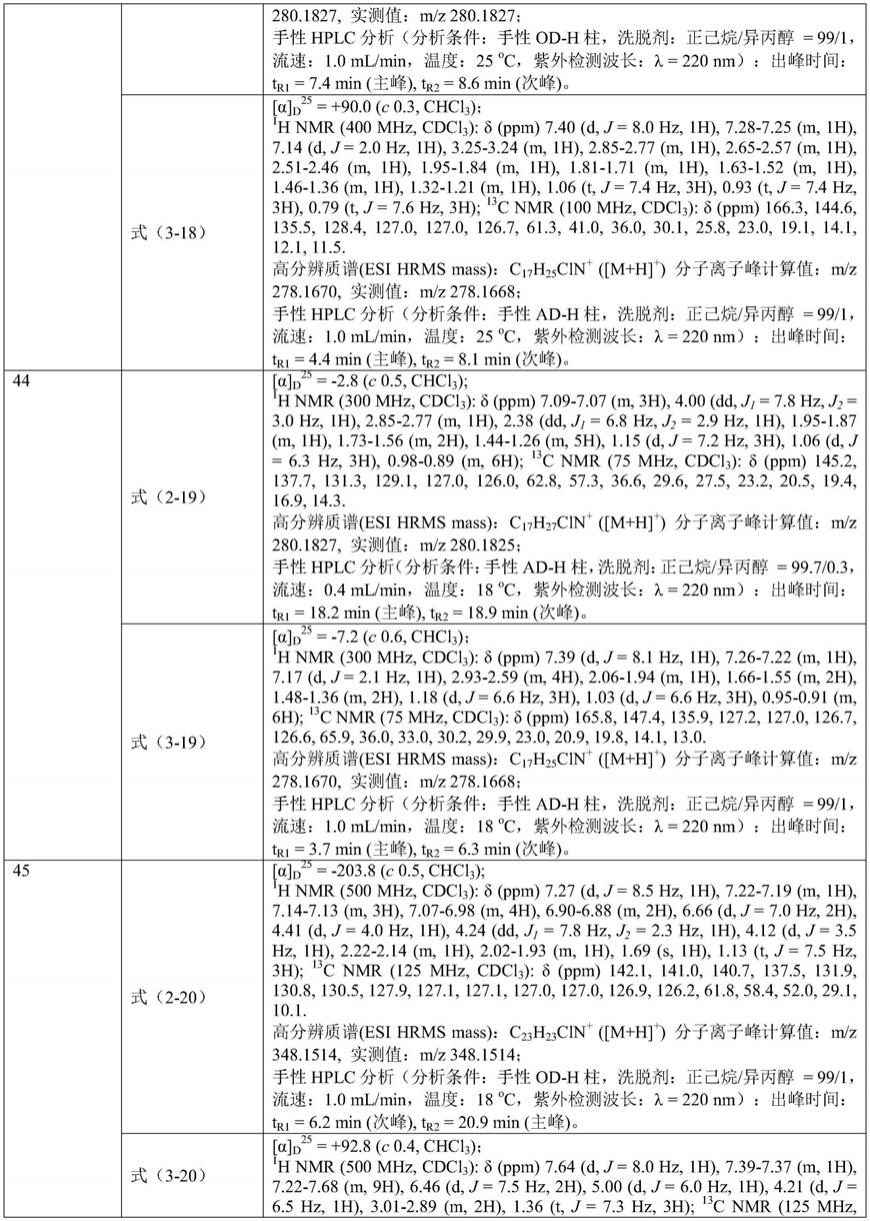
式(i-4)式(i-5)式(i-6)
[0050]
式(i-7)式(i-8)
[0051]
其中,ar1和ar2各自独立地选自取代或未取代的苯基、取代或未取代的萘基;且ar1和ar2中任选存在的取代基各自独立地选自c1-c3的烷基、c1-c3的烷氧基、c1-c3的羟烷基、卤素原子、羟基和羧基中的至少一种;
[0052]
r选自c1-c8的烷基、三氟甲基、取代或未取代的苯基、取代或未取代的萘基;且r中任选存在的取代基各自独立地选自c1-c8的烷基、甲氧基、氟、氯、溴、硝基和三氟甲基中的至少一种;优选地,r为甲基、对甲基苯基、对三氟甲基苯基、萘基、2,3,4-三异丙基苯基或三氟甲基;
[0053]
r'选自c1-c10的烷基、三氟甲基、取代的胺基、取代或未取代的苯基、取代或未取代的萘基;且r'中任选存在的取代基各自独立地选自c
1-10
的烷基、甲氧基、氟、氯、溴、硝基和三氟甲基中的至少一种;优选地,r'为甲基、对甲基苯基、萘基、2,3,4-三异丙基苯基或三氟甲基;
[0054]
r”选自氢、取代或未取代的苄基或c1-c10的烷基;且r”中任选存在的取代基各自独立地选自c1-c10的烷基、甲氧基、氟、氯、溴、硝基和三氟甲基中的至少一种;优选为h。
[0055]
上述式(i-1)所示结构的化合物为(r,r)-n-单磺酰-二芳基乙二胺类化合物,作为这类化合物的实例包括:
[0056]
式(r,r)-(i-1-1-1)式(r,r)-(i-1-1-2)
[0057]
式(r,r)-(i-1-1-3)式(r,r)-(i-1-1-4)
[0058]
式(r,r)-(i-1-1-5)
[0059]
式(r,r)-(i-1-1-6)式(r,r)-(i-1-1-7)
[0060]
式(r,r)-(i-1-1-8)
[0061]
上述式(i-2)所示结构的化合物为(r,r)-n-单磺酰-环己二胺类化合物,作为这类化合物的实例包括:式(r,r)-(i-2-1-1)式(r,r)-(i-2-1-2)
[0062]
式(i-3)所示结构的化合物为(r,r)-n-单磺酰-1-取代吡咯-3,4-二胺类化合物,式(i-4)所示结构的化合物为(r)-n-单磺酰-2,2'-二氨基-1,1'-联萘二胺类化合物。
[0063]
上述式(i-5)所示结构的化合物为(s,s)-n-单磺酰-二芳基乙二胺类化合物,作为这类化合物的实例包括:式(s,s)-(i-5-1-1)式(s,s)-(i-5-1-2)
式(s,s)-(i-5-1-3)式(s,s)-(i-5-1-4)式(s,s)-(i-5-1-5)式(s,s)-(i-5-1-6)(s,s)-(i-5-1-7)
[0064]
上述式(i-6)所示结构的化合物为(s,s)-n-单磺酰-环己二胺类化合物,作为这类化合物的实例包括:式(s,s)-(i-6-1-1)式(s,s)-(i-6-1-2)
[0065]
式(i-7)所示结构的化合物为(s,s)-n-单磺酰-1-取代吡咯-3,4-二胺类化合物,式(i-8)所示结构的化合物为(s)-n-单磺酰-2,2'-二氨基-1,1'-联萘二胺类化合物
[0066]
根据本发明,上述式(4)所示结构的配合物中,作为另一配位基的l1为金属元素m提供了6配位或5配位的空间配位结构,这样的配位能够有助于式(4)所示结构的配合物具有较高的化学稳定性,从而有助于其作为本发明的手性催化剂时能够发挥高效、高对映选择性的催化作用,优选情况下,l1为η
6-苯配位基、η
6-1,4-二甲基苯配位基、η
6-1-甲基-4-异丙基苯配位基、η
6-1,3,5,-三甲基苯配位基、η
6-1,2,3,4,5-五甲基苯配位基、η
6-1,2,3,4,5,6-六甲基苯配位基、η
5-茂配位基或η
5-五甲基茂配位基,更优选为η
6-1-甲基-4-异丙基苯配位基或η
6-1,2,3,4,5,6-六甲基苯配位基。
[0067]
根据本发明,上述式(4)所示结构的配合物中,负离子x为的:otf-指的是三氟甲酸负离子,bf
4-指的是四氟化硼负离子,pf
6-指的是六氟化磷负离子,sbf
6-指的是六氟化锑负离子,ntf
2-指的是二(三氟甲磺酰)亚胺负离子,二芳基磷酸负离子例如可以为优选所述二芳基磷酸负离子具有式所示的结构;并以bar
4-表示四芳基硼负离子;
[0068]
其中,负离子x为的四芳基硼负离子中的芳基例如可以为取代或未取代的苯基或者取代或未取代的萘基,取代基为甲基、乙基、卤素或三氟甲基,优选地,负离子x为的四芳
基硼负离子中的芳基为苯基或3,5-二(三氟甲基)苯基;并以barf-表示芳基为3,5-二(三氟甲基)苯基的四芳基硼负离子,以bph
4-表示芳基为苯基的四芳基硼负离子。优选地,x为的联芳基二酚衍生的磷酸负离子为下式所示结构中的一种:
[0069][0070][0071]
上述式(ii-1)所示结构的是二苯基磷酸负离子,式(ii-2)所示结构的是2,2'-联苯磷酸负离子,式(ii-3)所示结构的是(r)-2,2'-联二萘磷酸负离子,式(ii-4)所示结构的是(s)-2,2'-联二萘磷酸负离子,式(ii-5)所示结构的是(r)-8h-2,2'-联二萘磷酸负离子,式(ii-6)所示结构的是(s)-8h-2,2'-联二萘磷酸负离子。
[0072]
为了获得更高产率地和/或更高对映体对量地制得多取代手性四氢异喹啉类化合物和多取代手性二氢异喹啉类化合物的单一光学异构体,更优选地,x为otf-、bf
4-、pf
6-、sbf
6-、barf-或式(ii-1)所示的结构。
[0073]
根据本发明,典型的手性催化剂如下式所示的结构:
[0074]
[0075]
[0076][0077]
其中:定义x=otf-(a),bf
4-(b),pf
6-(c),sbf
6-(d),ntf
2-(e),barf-(f),二苯基磷酸负离子(g),2,2
’-
联苯磷酸负离子(h),(r)-2,2
’-
联二萘磷酸负离子(i),(s)-2,2
’-
联二萘磷酸负离子(j)。
[0078]
即,在本文中,(r,r)-4a指的是具有上述(r,r)-4的结构且其中x=otf。也即,上述典型的手性催化剂选自以下化合物中的一种或多种:(r,r)-4a、(r,r)-4b、(r,r)-4c、(r,r)-4d、(r,r)-4e、(r,r)-4f、(r,r)-4g、(r,r)-4h、(r,r)-4i、(r,r)-4j、(r,r)-5a、(r,r)-5b、(r,r)-5c、(r,r)-5d、(r,r)-5e、(r,r)-5f、(r,r)-5g、(r,r)-5h、(r,r)-5i、(r,r)-5j、(r,r)-6a、(r,r)-6b、(r,r)-6c、(r,r)-6d、(r,r)-6e、(r,r)-6f、(r,r)-6g、(r,r)-6h、(r,r)-6i、(r,r)-6j、(r,r)-7a、(r,r)-7b、(r,r)-7c、(r,r)-7d、(r,r)-7e、(r,r)-7f、(r,r)-7g、(r,r)-7h、(r,r)-7i、(r,r)-7j、(r,r)-8a、(r,r)-8b、(r,r)-8c、(r,r)-8d、(r,r)-8e、(r,r)-8f、(r,r)-8g、(r,r)-8h、(r,r)-8i、(r,r)-8j、(r,r)-9a、(r,r)-9b、(r,r)-9c、(r,r)-9d、(r,r)-9e、(r,r)-9f、(r,r)-9g、(r,r)-9h、(r,r)-9i、(r,r)-9j、(r,r)-10a、(r,r)-10b、(r,r)-10c、(r,r)-10d、(r,r)-10e、(r,r)-10f、(r,r)-10g、(r,r)-10h、(r,r)-10i、(r,r)-10j、(r,r)-11a、(r,r)-11b、(r,r)-11c、(r,r)-11d、(r,r)-11e、(r,r)-11f、(r,r)-11g、(r,r)-11h、(r,r)-11i、(r,r)-11j、(r,r)-12a、(r,r)-12b、(r,r)-12c、(r,r)-12d、(r,r)-12e、(r,r)-12f、(r,r)-12g、(r,r)-12h、(r,r)-12i、(r,r)-12j、(r,r)-13a、(r,r)-13b、(r,r)-13c、(r,r)-13d、(r,r)-13e、(r,r)-13f、(r,r)-13g、(r,r)-13h、(r,r)-13i、(r,r)-13j、(r,r)-14a、(r,r)-14b、(r,r)-14c、(r,r)-14d、(r,r)-14e、(r,r)-14f、(r,r)-14g、(r,r)-14h、(r,r)-14i、(r,r)-14j、(r,r)-15a、(r,r)-15b、(r,r)-15c、(r,r)-15d、(r,r)-15e、(r,r)-15f、(r,r)-15g、(r,r)-15h、(r,r)-15i、(r,r)-15j、(r,r)-16a、(r,r)-16b、(r,r)-16c、(r,r)-16d、(r,r)-16e、(r,r)-16f、(r,r)-16g、(r,r)-16h、(r,r)-16i、(r,r)-16j、(r,r)-17a、(r,r)-17b、(r,r)-17c、(r,r)-17d、(r,r)-17e、(r,r)-17f、(r,r)-17g、(r,r)-17h、(r,r)-17i、(r,r)-17j、18a、18b、18c、18d、18e、18f、18g、18h、18i、18j、(s,s)-4a、(s,s)-4b、(s,s)-4c、(s,s)-4d、(s,s)-4e、(s,s)-4f、(s,s)-4g、(s,s)-4h、(s,s)-4i、(s,s)-4j、(s,s)-5a、(s,s)-5b、(s,s)-5c、(s,s)-5d、(s,s)-5e、(s,s)-5f、(s,s)-5g、(s,s)-5h、(s,s)-5i、(s,s)-5j、(s,s)-6a、(s,s)-6b、(s,s)-6c、(s,s)-6d、(s,s)-6e、(s,s)-6f、(s,s)-6g、
(s,s)-6h、(s,s)-6i、(s,s)-6j、(s,s)-7a、(s,s)-7b、(s,s)-7c、(s,s)-7d、(s,s)-7e、(s,s)-7f、(s,s)-7g、(s,s)-7h、(s,s)-7i、(s,s)-7j、(s,s)-8a、(s,s)-8b、(s,s)-8c、(s,s)-8d、(s,s)-8e、(s,s)-8f、(s,s)-8g、(s,s)-8h、(s,s)-8i、(s,s)-8j、(s,s)-9a、(s,s)-9b、(s,s)-9c、(s,s)-9d、(s,s)-9e、(s,s)-9f、(s,s)-9g、(s,s)-9h、(s,s)-9i、(s,s)-9j、(s,s)-10a、(s,s)-10b、(s,s)-10c、(s,s)-10d、(s,s)-10e、(s,s)-10f、(s,s)-10g、(s,s)-10h、(s,s)-10i、(s,s)-10j、(s,s)-11a、(s,s)-11b、(s,s)-11c、(s,s)-11d、(s,s)-11e、(s,s)-11f、(s,s)-11g、(s,s)-11h、(s,s)-11i、(s,s)-11j、(s,s)-12a、(s,s)-12b、(s,s)-12c、(s,s)-12d、(s,s)-12e、(s,s)-12f、(s,s)-12g、(s,s)-12h、(s,s)-12i、(s,s)-12j、(s,s)-13a、(s,s)-13b、(s,s)-13c、(s,s)-13d、(s,s)-13e、(s,s)-13f、(s,s)-13g、(s,s)-13h、(s,s)-13i、(s,s)-13j、(s,s)-14a、(s,s)-14b、(s,s)-14c、(s,s)-14d、(s,s)-14e、(s,s)-14f、(s,s)-14g、(s,s)-14h、(s,s)-14i、(s,s)-14j、(s,s)-15a、(s,s)-15b、(s,s)-15c、(s,s)-15d、(s,s)-15e、(s,s)-15f、(s,s)-15g、(s,s)-15h、(s,s)-15i、(s,s)-15j、(s,s)-16a、(s,s)-16b、(s,s)-16c、(s,s)-16d、(s,s)-16e、(s,s)-16f、(s,s)-16g、(s,s)-16h、(s,s)-16i、(s,s)-16j、(s,s)-17a、(s,s)-17b、(s,s)-17c、(s,s)-17d、(s,s)-17e、(s,s)-17f、(s,s)-17g、(s,s)-17h、(s,s)-17i、(s,s)-17j。上述化合物更优选选择x=otf-(a),bf
4-(b),pf
6-(c),sbf
6-(d),ntf
2-(e),barf-(f),二苯基磷酸负离子(g),2,2
’-
联苯磷酸负离子(h),(r)-2,2
’-
联二萘磷酸负离子(i),(s)-2,2
’-
联二萘磷酸负离子(j)时的所示结构的配合物作为手性催化剂。从提高本发明的式(1)所示结构的化合物的氢化转化率方面考虑,更优选地,采用x=otf-(a),bf
4-(b),pf
6-(c),sbf
6-(d),barf-(f),二苯基磷酸负离子(g)或2,2
’-
联苯磷酸负离子(h)的上述配合物作为本发明的手性催化剂。从同时提高本发明的式(1)所示结构的化合物氢化转化率和对映体过量值方面考虑,更优选地,采用x=二苯基磷酸负离子(g)的上述配合物作为本发明的催化剂(特别优选采用(r,r)-4g、(r,r)-5g,及其对映体(s,s)-4g、(s,s)-5g中的至少一种)。
[0079]
所述手性二胺金属催化剂优选采用cn105111208a中公开的手性催化剂制备方法制备,本发明对此并无特别的限定。
[0080]
根据本发明,采用式(4)所示结构的配合物作为式(1)所示结构的化合物与氢气进行加成反应的催化剂,若使用手性催化剂即可高产率和高对映体过量地获得式(2)、式(3)所示结构的化合物。然而根据式(1)所示结构的化合物的结构特点,为了更为优化所述手性催化剂对式(1)所示结构的化合物的催化活性,优选情况下,式(1)所示结构的化合物与所述手性催化剂的用量的摩尔比为10-2000:1,例如可以为10-30:1、20-40:1、30-50:1、45-100:1、50-150:1、50-200:1、100-250:1、100-300:1、350-400:1、450-500:1、500-1000:1或500-1500:1,更优选为50-1000:1,更进一步优选为50-500:1。
[0081]
上述与氢气加成反应可以采用本领域常规的氢气催化氢化反应的条件,但是为了更配合本发明的手性催化剂对底物的催化作用,优选情况下,所述加成反应的条件包括:氢气的压力为1-100atm,温度为-10至100℃,时间为0.5-72h。作为上述加成反应的条件的氢气的压力例如可以为10-100atm、30-100atm、50-100atm、80-100atm、30-80atm、30-50atm或50-80atm,更优选为5-80atm,更进一步优选为50-80atm。作为上述加成反应的条件的温度例如可以为-10至90℃、-10至60℃、-10至40℃、-10至25℃、25-90℃、25-60℃、25-40℃、40-90℃、40-60℃或60-90℃,更优选为0-60℃,更进一步优选为0-40℃。作为上述加成反应的
条件的时间例如可以为0.5-5h、5-10h、11-15h、16-20h或16-20h,更优选为0.5-15h,更进一步优选为0.5-5h。其中氢气的压力为1atm是指反应体系处于氢气的压力达到1atm的环境中。上述加成反应可以在多种反应容器中进行,优选采用高压反应釜进行。
[0082]
根据本发明,对所述加成反应采用的溶剂没有特别的限定,可以为水和常规的有机溶剂,例如可以为咪唑类离子液体[bmim]pf6、水、二氯甲烷(dcm)、1,2-二氯乙烷、氯仿、乙酸乙酯(ea)、四氢呋喃(thf)、苯、甲苯、二甲苯、氯代苯、乙醚、二氧六环、丙酮和c1-c10的一元醇中的一种或多种,其中,c1-c10的一元醇优选为甲醇(meoh)、乙醇(etoh)、丙醇、正丁醇(n-buoh)和异丙醇(ipa)中的一种或多种。更优选地,所述溶剂为甲醇(meoh)、乙醇(etoh)、异丙醇(ipa)、正丁醇(n-buoh)、二氯甲烷(dcm)、四氢呋喃(thf)、甲苯、乙酸乙酯(ea)和丙酮中的一种或多种。其中,采用甲醇(meoh)、乙醇(etoh)、异丙醇(ipa)、正丁醇(n-buoh)、二氯甲烷(dcm)、四氢呋喃(thf)、甲苯、乙酸乙酯(ea)或丙酮作为溶剂时,可以获得ee值为93%以上的式(2)所示结构的化合物和ee值为99%以上的式(3)所示结构的化合物。
[0083]
作为上述溶剂的另一种优选选择为将二氯甲烷(dcm)、1,2-二氯乙烷、四氢呋喃、苯、甲苯、二甲苯和氯代苯中的至少一种和c1-c10的一元醇的混合溶液作为溶剂。例如可以为体积比为1-2:1的异丙醇和甲苯的混合溶剂作为上述加成反应的溶剂;或者是,将体积比为1-2:1的异丙醇和二氯甲烷的混合溶剂作为上述加成反应的溶剂。
[0084]
根据本发明,对式(1)所示结构的化合物的用量没有特别的限定,只要能够获得本发明的式(2)、式(3)所示结构的化合物即可,优选情况下,相对于1ml的溶剂,式(1)所示结构的化合物的摩尔用量为0.1-1mmol,更优选为0.1-0.6mmol。
[0085]
本发明的方法能够将式(1)所示结构的化合物选择性地进行氢化动力学拆分,得到具有手性碳原子的所述多取代四氢异喹啉和相应的多取代二氢异喹啉的手性化合物。
[0086]
本发明第二方面提供了一种多取代手性四氢异喹啉类化合物,所述多取代手性四氢异喹啉类化合物为式(2)所示的化合物;
[0087]
式(2)各个基团如上文所描述的。
[0088]
本发明第三方面提供了一种多取代手性二氢异喹啉类化合物的单一光学异构体,其特征在于,所述多取代手性二氢异喹啉类化合物为式(1)所示的化合物;
[0089]
式(1)各个基团如上文所描述的。
[0090]
具体的化合物如上文中所描述的,这里不再赘述。
[0091]
本发明能够获得对映体过量高达93%的多取代手性四氢异喹啉,非对映异构体过量99%以上。其中,优选地,本发明能够获得对映体过量为99%以上的多取代手性二氢异喹
啉,非对映异构体过量99%以上。
[0092]
以下将通过实施例对本发明进行详细描述。
[0093]
以下例子中
[0094]
反应的转化率=[转化的反应物]/([转化的反应物]+[未转化的反应物])
×
100%。本发明式(1)所示结构的化合物的不对称催化氢化反应的转化率是将纯化前的不对称催化氢化反应产物混合物直接进行核磁共振氢谱(1h-nmr)分析,其中未反应的式(1)所示结构的化合物的特征峰的峰面积与已转换为产物的特征峰的峰面积分别看作未转化的反应物与转化的反应物的浓度(重量百分含量),根据上述公式进行计算得到转化率。
[0095]
对于具有两个手性中心的氢化产物来说,trans/cis指的是反应产物中非对映异构体的比值,计算公式为[(s,s)+(r,r)]/[(s,r)+(r,s)];其中,对于trans更多的产物来说,产物的对映体过量(ee值的绝对值,表示反应产物中一个对映体对另一个对映体的过量,通常用百分数表示)的计算公式为:ee=[(s,s)-(r,r)]/[(s,s)+(r,r)])
×
100%;而对于cis更多的产物来说,产物的对映体过量(ee值的绝对值)的计算公式为:ee=[(s,r)-(r,s)]/[(s,r)+(r,s)]
×
100%。
[0096]
以下手性催化剂均按照cn105111208a中公开的手性催化剂制备方法制备得到。
[0097]
式(1)所示结构的化合物均采用以下方法制得,由下述方法制得的均为式(1)所示结构的化合物的外消旋体:
[0098][0099]
将式(1-a)和式(1-b)溶于乙醚溶液(式(1-a)所示结构化合物和式(1-b)所示结构化合物的用量摩尔比为1:1.5)有机溶剂的用量使得式(1-a)所示结构的化合物的浓度为1.5mol/l,加入fe3(co)
12
(式(1-a)所示结构化合物和fe3(co)
12
的用量摩尔比为1:0.05),在氮气氛围下进行氧化还原中性的4+2环加成反应,反应完毕后冷却至室温,旋干后得到粗产物,用柱层析纯化得到式(1)所示结构的化合物。
[0100]
实施例1-13
[0101]
本实施例用于说明本发明的多取代二氢异喹啉类化合物对映异构体的混合物的不对称氢化动力学拆分方法。
[0102]
在高压反应釜中,将0.01mmol的手性催化剂(具体的选择见表1)、0.1mmol的式(1-1)所示结构的化合物外消旋体,溶解于1.0ml二氯甲烷中,用氮气置换空气后,充入50atm的氢气,并在25℃下搅拌反应一定时间,将反应后得到的反应液硅胶柱层析以除去手性催化剂,得到式(2-1)结构所示的手性多取代四氢异喹啉和式(3-1)结构所示的手性多取代二氢异喹啉,测试得到的化合物的ee值,ee值和转化率如表1所示。
[0103]
表1
[0104][0105]
实施例14-20
[0106]
本实施例用于说明本发明的多取代二氢异喹啉类化合物对映异构体的混合物的不对称氢化动力学拆分方法。
[0107]
在高压反应釜中,将0.01mmol手性催化剂(r,r)-4c、0.1mmol的式(1-1)所示结构的化合物外消旋体,溶解于1.0ml溶剂中(具体的选择见表2),用氮气置换空气后,充入50atm的氢气,并在25℃下搅拌反应一定时间,将反应后得到的反应液硅胶柱层析以除去手性催化剂,得到式(2-1)结构所示的手性多取代四氢异喹啉和式(3-1)结构所示的手性多取代二氢异喹啉,测试得到的化合物的ee值,ee值和转化率如表2所示。
[0108]
表2
[0109][0110]
实施例21-26
[0111]
本实施例用于说明本发明的的多取代二氢异喹啉类化合物对映异构体的混合物的不对称氢化动力学拆分方法。
[0112]
在高压反应釜中,将一定量的手性催化剂(r,r)-4c、0.1mmol的式(1-1)所示结构的化合物外消旋体,溶解于1.0ml二氯甲烷中,用氮气置换空气后,充入一定量的的氢气,并在一定的温度下搅拌反应一定时间,将反应后得到的反应液硅胶柱层析以除去手性催化剂,得到式(2-1)结构所示的手性多取代四氢异喹啉和式(3-1)结构所示的手性多取代二氢异喹啉,测试得到的化合物的ee值,ee值和收率如表3所示。
[0113]
表3
[0114][0115]
实施例27-48
[0116]
本实施例用于说明本发明的多取代二氢异喹啉类化合物对映异构体的混合物的不对称氢化动力学拆分方法。
[0117]
在高压反应釜中,将0.01mmol的手性催化剂(r,r)-4c和0.1mmol的指定的式(1)所示结构的化合物外消旋体溶解于1ml的二氯甲烷中,用氮气置换空气后,充入50atm的氢气,并在25℃下搅拌反应一定的时间,将所得反应液经硅胶柱层析(洗脱液为二氯甲烷)以除去手性催化剂。所采用的式(1)所示结构的消旋多取代二氢异喹啉衍生物的结构不同,其余均与实施例23相同,得到手性多取代四氢异喹啉化合物(2)和回收相应的手性多取代二氢异喹啉化合物(3),测试各实施例制备得到的手性化合物的ee值,ee值和收率具体如表4所示。
[0118]
表4
[0119][0120][0121]
为了直观的说明利用本发明的消旋多取代二氢异喹啉化合物的动力学拆分方法
制备得到的手性多取代四氢异喹啉化合物和相应的手性3,4-二氢异喹啉类化合物的性能及表征过程,本发明示例性地提供了实施例23和实施例31-52所制备的手性化合物的鉴定结果和过程,具体如表5所示。
[0122]
表5
[0123]
[0124]
[0125]
[0126]
[0127]
[0128]
[0129]
[0130]
[0131]
[0132]
[0133][0134]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1