喹唑啉衍生物及其作为抗肿瘤药物的应用的制作方法
本发明属于药物合成领域,具体地说是喹唑啉衍生物及其作为抗肿瘤药物的应用。
背景技术:
酪氨酸激酶抑制剂(tyrosinekinaseinhibitor),主要作用于表皮生长因子受体(epidermalgrowthfactorreceptor,egfr/her1)的激酶结构域,阻断细胞生长信号。表皮生长因子受体是人表皮生长因子受体(her)四大受体之一,其他三个分别是her2(erbb2)、her3(erbb3)和her4(erbb4)。egfr横跨细胞膜,由膜外配受体结合域,单个跨膜段和包含酪氨酸激酶结构域的胞内区。从结构来看,egfr是同时具备激酶活性的糖蛋白,它即作为受体,又作为酶,在细胞的增值,分化,凋亡等生理活动中扮演着重要角色。有证据表明肿瘤细胞的发生和转移常常伴随着her1(egfr)和her2的过表达。
非小细胞肺癌是目前已知的具有egfr突变特征的癌症之一,但是上市药物的耐药问题亟待解决,而且目前针对胃癌和肺癌治疗的靶点主要包括her1、her2、vegf、vegfr、hgf等,但临床疗效不佳,因此,需要研发一种抗耐药性、对胃癌和肺癌细胞株抑制活性强的喹唑啉衍生物。
技术实现要素:
发明目的:
本发明的目的是提供一种喹唑啉类衍生物和其盐类,尤其是其与无机的或有机的酸的生理上可接受的盐类,并提供含有该药理学上有效化合物的药物制剂及其应用。
技术方案:
本发明是通过以下技术方案来实现的。
通式(i)所示的喹唑啉衍生物或其药学上可接受的盐:
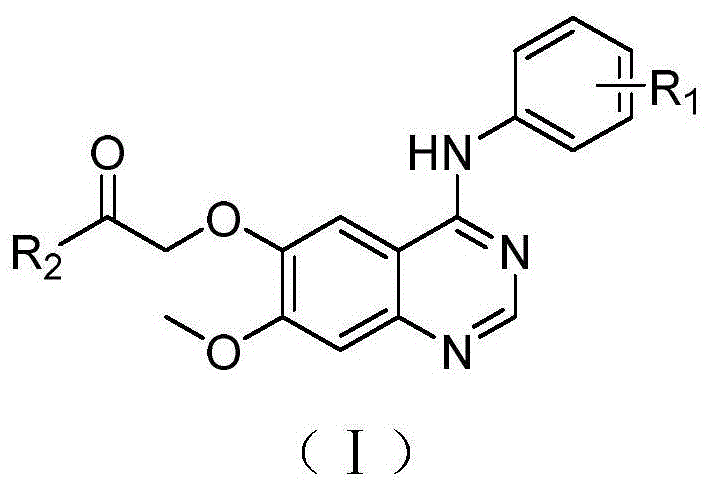
其中,r1表示:氢、c1-c3烷基、c1-c3烷氧基或卤素;r2表示:c1-c3仲氨基、取代的五元杂环氨基或取代的六元杂环氨基。
所述的喹唑啉衍生物或其可药用盐,r1选自单取代的氢、氟、甲基、甲氧基或二取代的甲基和氟;r2选自二甲氨基、二乙氨基或吗啉基。
所述的喹唑啉衍生物或其药学上可接受的盐,喹唑啉衍生物或其药学上可接受的盐与酸形成药用盐,适用的酸为:盐酸、氢溴酸、磷酸、硫酸、酒石酸、水杨酸、甲磺酸、琥珀酸、富马酸、丁二酸、乳酸、柠檬酸、苹果酸或马来酸。
所述任一化合物作为制备治疗以异常egfr家族疾病的药物中的应用。
所述任一化合物或盐类,以及含有该任一化合物或盐类作为活性成分的药物组合物作为制备抗肿瘤药物的应用。
所述任一化合物或盐类,以及含有该任一化合物或盐类作为活性成分的药物组合物的应用,其用于制备皮下给药或口服的药物制剂
优点及效果:
本发明的喹唑啉衍生物还具有但不限于以下有益效果:
(1)相对吉非替尼,本发明所提供的喹唑啉衍生物大部分具有良好的药理活性。其中,喹唑啉衍生物i-b、i-b″、i-c″和i-d″对h1975细胞生长的抑制活性ic50值分别为0.720、0.842、0.478和0.652μmol/l,优于阳性对照药吉非替尼的6.925μmol/l;喹唑啉衍生物i-b′、i-g′和i-f″对mkn45细胞生长的抑制活性ic50值分别为0.376、0.532和0.324μmol/l,优于阳性对照药吉非替尼的3.281μmol/l。因此,所设计的喹唑啉衍生物具有更好的抑制活性。
(2)本发明所提供的部分喹唑啉衍生物具有良好的选择性。其中,唑啉衍生物i-b′对体外培养mkn45和h1975细胞生长的抑制活性ic50值分别为0.376μmol/l和18.411μmol/l;唑啉衍生物i-d″对体外培养mkn45和h1975细胞生长的抑制活性ic50值分别为7.548μmol/l和0.6521μmol/l。由此可见,所设计的喹唑啉衍生物具有更好的选择性,化合物i-b′有利于对mkn45细胞的抑制,而化合物i-d″有利于对耐药细胞株h1975的抑制。
附图说明:
图1是喹唑啉衍生物或其药学上可接受的盐的通式;
图2是合成通式(i)所示的化合物衍生物的路线图。
具体实施方式:
本发明提供了一类抗耐药性较好的喹唑啉类衍生物。本发明提供了一系列对胃癌和肺癌细胞株抑制活性强的喹唑啉衍生物。
吗啉在药物修饰设计中被广泛应用,有研究表明在具有良好生物活性的分子骨架上引入吗啉基时,可以增强药物的药代动力学性质。如:抗肿瘤药物吉非替尼、卡奈替尼等。分子量较小的叔胺基常被用来作为药物活性分子的亲油性侧链,可以大大提高药物分子的生物活性。如:egf816、来那替尼等。
基于药物构效关系理论、生物电子等排原理、药代动力学原理等药物设计理念,本发明以4-芳胺基喹唑啉为母体结构,对其4-位和6-位进行结构改造和修饰,以期获得特异性高、选择性好、毒性低的先导化合物,进而得到抗耐药性的靶向抗肿瘤药物。
本发明涉及通式(i)的化合物和其盐类、及其制法,含有该药理学上有效化合物的药物制剂及其应用。
如图1所示,通式(i)所示的喹唑啉衍生物或其药学上可接受的盐:
其中,r1表示:氢、c1-c3烷基、c1-c3烷氧基或卤素;r2表示:c1-c3仲氨基、取代的五元杂环氨基或取代的六元杂环氨基。
所述的喹唑啉衍生物或其可药用盐,r1选自单取代的氢、氟、甲基、甲氧基或二取代的甲基和氟;r2选自二甲氨基、二乙氨基或吗啉基。
所述的喹唑啉衍生物或其药学上可接受的盐,喹唑啉衍生物或其药学上可接受的盐与酸形成药用盐,适用的酸为:盐酸、氢溴酸、磷酸、硫酸、酒石酸、水杨酸、甲磺酸、琥珀酸、富马酸、丁二酸、乳酸、柠檬酸、苹果酸或马来酸。
所述任一化合物作为制备治疗以异常egfr家族疾病的药物中的应用。
所述任一化合物或盐类,以及含有该任一化合物或盐类作为活性成分的药物组合物作为制备抗肿瘤药物的应用。
所述任一化合物或盐类,以及含有该任一化合物或盐类作为活性成分的药物组合物的应用,其用于制备皮下给药或口服的药物制剂
本发明所述的化合物或其可药用盐可以单独或以药物组合物的形式给药。本发明药物组合物可根据给药途径配成各种适宜剂型。使用一种或多种生理学上可接受的载体,包含赋形剂和助剂,它们有利于将活性化合物加工成可以在药学上使用的制剂。适当的制剂形式取决于所选择的给药途径,可以按照本领域熟知的常识进行制备。
给药途径可以是口服、非肠道或局部给药,优选口服和注射形式给药。可以口服的药物制剂包括胶囊剂和片剂等。本发明化合物也可以配制用于肠胃外给药或者透皮给药或者经粘膜给药,或者采用栓剂或者埋植剂的方式给药。本领域技术人员可以理解,本发明化合物可以采用合适的药物释放系统(dds)以得到更有利的效果。
本发明通过体外四氮唑盐还原法(mtt法)试验表明:具有通式(i)结构的喹唑啉衍生物对人胃癌细胞(mkn45)、人肺腺癌细胞(a549)、人肺癌耐药细胞(h1975)等具有很强的细胞增殖抑制作用。
下面结合具体实施例对本发明做进一步的说明:
合成式(i)所示的化合物衍生物如下:
其中r1和r2如上所定义。
如图2所示,以3,4-二氢-7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯1为原料,甲苯为溶剂,三乙胺为缚酸剂,采用三氯氧磷为氯代试剂,先生成中间体4-氯-7-甲氧基喹唑啉-6-醇乙酸酯2;然后与取代的苯胺在加热条件下,发生亲核取代反应,得到中间体4-取代苯氨基-7-甲氧基喹唑啉-6-基乙酸酯3。在室温条件下,用甲醇溶解上述中间体,缓慢滴加氨水进行酯的水解反应,生成中间体4-取代苯氨基-7-甲氧基-6-羟基喹唑啉4。在氮气保护条件下,以甲苯或二氯甲烷为溶剂,三乙胺作为缚酸剂,采用吗啉、二甲胺、二乙胺与氯乙酰氯5发生酰胺化反应生成中间体侧链6。最后,酰胺中间体侧链6与关键中间体4-取代苯氨基-7-甲氧基-6-羟基喹唑啉4发生醚化反应生成目标衍生物i。
实施例1
2-(7-甲氧基-4-苯胺基)喹唑啉-6-基)-1-吗啉-1-酮(i-a)的合成;
以甲苯(30ml)作为溶剂,将3,4-二氢-7-甲氧基-4-氧代喹唑啉-6-醇乙酸酯1(3.0g,12.81mmol)、三乙胺(3.1ml,21.78mmol)和三氯氧磷(3ml,32.02mmol)依次加入至150ml单口瓶内,在78℃恒温搅拌回流条件下,反应3h左右。tlc监测反应进度(乙酸乙酯/石油醚=1/1)。待原料完全反应后,瓶内悬浊液不经处理,直接进行第二步反应。向上述悬浊液中缓慢滴入用甲苯(40ml)稀释的苯胺a(1.55g,16.67mmol)溶液,继续搅拌4h,产生大量固体。tlc监测(乙酸乙酯/石油醚=2/1)。反应完毕,反应液降至室温后减压抽滤,用甲苯(20ml)润洗,干燥,得淡黄色粉末固体,再用异丙醇(20ml)重结晶,干燥得类白色粉末状固体3a3.40g。两步总收率85.7%;ir(v,cm-1):2748,2117,1777,1628,1560,1427,1217,1153,1064,991,843,745,578,493;1h-nmr(dmso-d6,400mhz):δ11.64(s,1h),8.89(d,j=15.2hz,2h),7.71(d,j=8.0hz,2h),7.59(s,1h),7.49(t,j=7.6hz,2h),7.33(t,j=7.2hz,1h),4.01(s,3h),2.39(s,3h).
将中间体3a(1.5g,4.85mmol)与甲醇(10ml)混合于50ml单口瓶内,低温搅拌降至0℃后,加入氨水溶液(8ml,浓度25-28%),投料完毕,升至室温搅拌2~4h。tlc监测反应(二氯甲烷/甲醇=30/1),反应完毕,得米白色悬浊液。减压抽滤,用少量甲醇水溶液洗涤,干燥,得白色偏黄粉末状固体中间体4a1.11g,收率85.6%;ir(v,cm-1):3390,2346,2116,1832,1605,1579,1521,1443,1243,1067,956,840,741,485;1h-nmr(400mhz,dmso-d6):δ9.37(s,1h,nh),8.43(s,1h,arh),7.89–7.70(m,3h,arh),7.36(t,j=7.6hz,2h,arh),7.20(s,1h,arh),7.07(t,j=7.2hz,1h,arh),3.98(s,3h,ch3).
将吗啉x(1.21g,13.70mmol)、三乙胺(1.38g,13.67mmol)和甲苯(10ml)混合于50ml单口瓶中,在-5℃冰浴和氮气保护条件下搅拌,缓慢加入氯乙酰氯(1.56g,13.81mmol)于上述液体中。1h后,将反应转移到室温,并在氮气保护下反应12h。tlc监测反应(二氯甲烷/乙酸乙酯=35/1),反应完全后,用饱和氯化铵水溶液(30ml)洗2次(15ml×2)。有机相再次用去离子水(15ml)清洗,无水硫酸镁干燥,过滤,浓缩得黄色粘稠液体2-氯-1-吗啉乙酮6x1.89g,收率:83.18%。将侧链中间体密封,于阴凉干燥处保存留以备用。
将中间体4a(0.1g,0.374mmol)、2-氯-1-吗啉乙酮6x(0.093g,0.568mmol)和碳酸钾(0.103g,0.748mmol)混合溶于30ml干燥的n,n-二甲基甲酰胺中,在80℃下搅拌3h,tlc监测(二氯甲烷/甲醇=15/1)。反应毕,将混合物冷却至室温,然后缓慢加入冷水并搅拌直至析出大量絮状固体。减压抽滤,干燥得灰白色粉末固体2-((7-甲氧基-4-(苯基氨基)喹唑啉-6-基)氧基)-1-吗啉-1-酮(i-a)0.092g,收率66%;m.p.:207.2~209.5℃;ir(v,cm-1):3348.73,3014.06,2962.86,2904.98,2860.98,1619.65,1361.39,1202.87,848.40;1h-nmr(600mhz,dmso-d6):δ9.39(s,1h,nh),8.46(s,1h,c=nh),7.85–7.76(m,3h,arh),7.40(t,j=7.2hz,2h,arh),7.22(s,1h,arh),7.12(t,j=6.8hz,1h,arh),4.98(s,2h,och2c=o),3.95(s,3h,och3),3.67(s,2h,och2),3.59(s,2h,och2),3.54(s,2h,nch2),3.50(s,2h,nch2)。
实施例2
2-(4-(2-氟苯基)氨基)-7-甲氧基喹唑啉-6-基)-1-吗啉-1-酮(i-b)的合成;
按照实施例1相同的方法制备,将原料苯胺替换为邻氟苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-b)0.109g,收率73%;m.p.:105.7~106.6℃;ir(v,cm-1):3293.84,2918.66,2854.35,1640.20,1504.79,1424.69,1206.89,998.80,844.15;1h-nmr(600mhz,dmso-d6):δ9.38(s,1h,nh),8.36(s,1h,c=nh),7.78(s,1h,arh),7.57(s,1h,arh),7.32(d,j=7.3hz,2h,arh),7.26(s,1h,arh),7.22(s,1h,arh),4.96(s,2h,och2c=o),3.95(s,3h,och3),3.66(s,2h,och2),3.60(s,2h,och2),3.53(s,2h,nch2),3.50(s,2h,nch2)。
实施例3
2-(4-(4-氟-2-甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-1-吗啉-1-酮(i-c)的合成;
按照实施例1相同的方法制备,将原料苯胺替换为4-氟-2-甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-c)0.118g,收率77%;m.p.:98.5~100.9℃;ir(v,cm-1):3397.98,3005.33,2969.50,2913.54,1650.50,1418.90,1229.24,997.61,839.55;1h-nmr(600mhz,dmso-d6):δ9.19(s,1h,nh),8.25(s,1h,c=nh),7.73(s,1h,arh),7.31(s,1h,arh),7.15(d,j=7.0hz,2h,arh),7.05(s,1h,arh),4.91(s,2h,och2c=o),3.91(s,3h,och3),3.62(s,2h,och2),3.56(s,2h,och2),3.49(s,2h,nch2),3.46(s,2h,nch2),2.14(s,3h,arch3)。
实施例4
2-(7-甲氧基-4-(4-甲氧基苯基)氨基)喹唑啉-6-基)-1-吗啉-1-1酮(i-d)的合成;
按照实施例1相同的方法制备,将原料苯胺替换为对甲氧基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-d)0.106g,收率73%;m.p.:140.3~142.1℃;ir(v,cm-1):3348.65,2965.30,2927.72,2864.36,1621.04,1583.54,1339.90,1004.54,847.94;1h-nmr(600mhz,dmso-d6):δ9.32(s,1h,nh),8.40(s,1h,c=nh),7.80(s,1h,arh),7.62(d,j=8.7hz,2h,arh),7.19(s,1h,arh),6.98(d,j=8.3hz,2h,arh),4.96(s,2h,och2c=o),3.95(s,3h,och3),3.78(s,3h,ch3),3.67(s,2h,och2),3.60(s,2h,och2),3.54(s,2h,nch2),3.50(s,2h,nch2)。
实施例5
2-(7-甲氧基-4-(3-甲氧基苯基)氨基)喹唑啉-6-基)-1-吗啉-1-酮(i-e)的合成;
按照实施例1相同的方法制备,将原料苯胺替换为间甲氧基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-e)0.107g,收率74%;m.p.:108.0~110.3℃;ir(v,cm-1):3326.99,2914.51,2835.88,1624.25,1576.39,1427.81,1208.95,1031.29,846.26;1h-nmr(600mhz,dmso-d6):δ9.35(s,1h,nh),8.50(s,1h,c=nh),7.82(s,1h,arh),7.50(s,1h,arh),7.39(t,j=10.0hz,1h,arh),7.30(t,j=8.1hz,1h,arh),7.23(s,1h,arh),6.71(d,j=8.1hz,1h,arh),4.99(s,2h,och2c=o),3.96(s,3h,och3),3.79(s,3h,ch3),3.68(s,2h,och2),3.60(s,2h,och2),3.55(s,2h,nch2),3.50(s,2h,nch2)。
实施例6
2-(4-(4-甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-1-吗啉-1-酮(i-f)的合成;
按照实施例1相同的方法制备,将原料苯胺替换为对甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-f)0.102g,收率77%;m.p.:167.7~168.9℃;ir(v,cm-1):3353.67,2963.83,2926.32,2865.33,1622.31,1514.41,1426.46,1239.55,821.44;1h-nmr(600mhz,dmso-d6):δ9.33(s,1h,nh),8.44(s,1h,c=nh),7.82(s,1h,arh),7.65(d,j=8.3hz,2h,arh),7.21(s,j=4.0hz,3h,arh),4.97(s,2h,och2c=o),3.95(s,3h,och3),3.68(s,2h,och2),3.60(s,2h,och2),3.54(s,2h,nch2),3.50(s,2h,nch2),2.31(s,3h,arch3)。
实施例7
2-(4-(2,4-二甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-1-吗啉-1-酮(i-g)的合成;
按照实施例1相同的方法制备,将原料苯胺替换为2,4-二甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-g)0.098g,收率68%;m.p.:119.1~120.4℃;ir(v,cm-1):3284.07,2974.16,2915.20,2863.63,1619.78,1504.04,1227.86,1010.49,850.22;1h-nmr(600mhz,dmso-d6):δ9.20(s,1h,nh),8.27(s,1h,c=nh),7.78(s,1h,arh),7.20–7.18(m,2h,arh),7.13(s,1h,arh),7.07(d,j=7.8hz,1h,arh),4.95(s,2h,och2c=o),3.95(s,3h,och3),3.67(s,2h,och2),3.60(s,2h,och2),3.53(s,2h,nch2),3.50(s,2h,nch2),2.32(s,3h,arch3),2.14(s,3h,arch3)。
实施例8
2-(7-甲氧基-4-(苯胺基)喹唑啉-6-基)氧二甲基乙酰胺(i-a′)的合成;
按照实施例1相同的方法制备到中间体4,然后将二甲胺y(1.05g,22.18mmol),三乙胺(4.49g,44.36mmol)和二氯甲烷(10ml)混合于50ml单口瓶中,在-5℃冰浴和氮气保护条件下搅拌,缓慢加入氯乙酰氯(2.53g,22.40mmol)于上述液体中。1h后,将反应转移到室温,并在氮气保护下反应12h。tlc监测反应(二氯甲烷/乙酸乙酯=35/1),反应完全后,用饱和氯化铵水溶液(30ml)洗2次(15ml×2)。有机相再次用去离子水(15ml)清洗,无水硫酸镁干燥,过滤,浓缩得黄色透明液体2-氯-n,n-二甲基乙酰胺6y2.51g,收率93.09%。将侧链中间体密封,于阴凉干燥处保存留以备用。
将中间体4a(0.19g,0.66mmol)、2-氯-n,n-二甲基乙酰胺6y(0.121g,0.99mmol)和碳酸钾(0.184g,1.33mmol)混合溶于30ml干燥的n,n-二甲基甲酰胺中,在80℃下搅拌3h,tlc监测(二氯甲烷/甲醇=15/1)。反应毕,将混合物冷却至室温,然后缓慢加入冷水并搅拌,直至析出大量絮状固体。减压抽滤,干燥得黄色粉状固体2-(7-甲氧基-4-(苯胺基)喹唑啉-6-基)氧二甲基乙酰胺(i-a′)0.155g,收率65%;m.p.:125.3~126.2℃;ir(v,cm-1):3367.73,2978.19,2945.41,1649.61,1474.44,1035.65,807.15;1h-nmr(400mhz,dmso-d6):δ9.39(s,1h,nh),8.45(s,1h,c=nh),7.75(s,1h,arh),7.43–7.32(m,3h,arh),7.24–7.05(m,3h,arh),4.96(s,2h,och2c=o),3.95(s,3h,och3),3.07(s,3h,nch3),2.88(s,3h,nch3)。
实施例9
2-(4-((2-氟苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二甲基乙酰胺(i-b′)的合成;
按照实施例8相同的方法制备,将原料苯胺替换为邻氟苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-b′)0.167g,收率67.70%;m.p.:100.2~101.9℃;ir(v,cm-1):3305.68,2930.98,1647.85,1500.15,1420.37,1138.96,853.55;1h-nmr(400mhz,dmso-d6):δ9.39(s,1h,nh),8.34(s,1h,c=nh),7.72(s,1h,arh),7.54(t,j=7.6hz,1h,arh),7.35–7.29(m,2h,arh),7.27(d,j=7.5hz,1h,arh),7.21(s,1h,arh),4.95(s,2h,och2c=o),3.95(s,3h,och3),3.05(s,3h,nch3),2.88(s,3h,nch3)。
实施例10
2-(4-((4-氟-2-甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二甲基乙酰胺(i-c′)的合成;
按照实施例8相同的方法制备,将原料苯胺替换为4-氟-2-甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-c′)0.171g,收率66%;m.p.:104.5~106.1℃;ir(v,cm-1):3344.42,2919.99,1646.09,1580.89,1421.40,1211.71,841.33;1h-nmr(400mhz,dmso-d6):δ9.23(s,1h,nh),8.27(s,1h,c=nh),7.70(s,1h,arh),7.37–7.31(m,1h,arh),7.19(d,j=8.8hz,2h,arh),7.09(t,j=7.3hz,1h,arh),4.93(s,2h,och2c=o),3.94(s,3h,och3),3.05(s,3h,nch3),2.88(s,3h,nch3),2.17(s,3h,arch3)。
实施例11
2-(4-((4-甲氧基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二甲基乙酰胺(i-d′)的合成;
按照实施例8相同的方法制备,将原料苯胺替换为对甲氧基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-d′)0.170g,收率63%;m.p.:72.7~73.5℃;ir(v,cm-1):3307.07,2928.94,2837.87,1650.73,1231.83,1029.62,844.96;1h-nmr(400mhz,dmso-d6):δ9.32(s,1h,nh),8.38(s,1h,c=nh),7.75(s,1h,arh),7.59(d,2h,arh,j=7.8hz),7.18(s,1h,arh),6.97(d,2h,j=7.7hz,arh),4.94(s,och2c=o),3.94(s,och3),3.77(s,och3),3.06(s,nch3),2.88(s,nch3)。
实施例12
2-(4-((3-甲氧基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二甲基乙酰胺(i-e′)的合成;
按照实施例8相同的方法制备,将原料苯胺替换为间甲氧基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-e′)0.172g,收率64%;m.p.:111.9~113.3℃;ir(v,cm-1):3358.03,2921.84,2832.86,1648.07,1244.66,1000.80,841.12;1h-nmr(400mhz,dmso-d6):δ9.32(s,1h,nh),8.38(s,1h,c=nh),7.75(s,1h,arh),7.59(d,2h,j=7.8hz,arh),6.97(d,3h,arh,j=7.7hz),4.94(s,2h,och2c=o),3.94(s,3h,och3),3.77(s,3h,och3),3.06(s,3h,nch3),2.88(s,3h,nch3)。
实施例13
2-(4-((4-甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二甲基乙酰胺(i-f′)的合成;
按照实施例8相同的方法制备,将原料苯胺替换为对甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-f′)0.175g,收率67%;m.p.:102.8~103.4℃;ir(v,cm-1):3309.47,2918.61,1647.31,1510.42,1425.39,1110.28,808.29;1h-nmr(400mhz,dmso-d6):δ9.36(s,1h,nh),8.47(s,1h,c=nh),7.78(s,1h,arh),7.47(s,1h,arh),7.36(d,1h,j=8.1hz,arh),7.30(d,1h,j=8.0hz,arh),7.21(s,1h,arh),6.71(d,1h,j=6.7hz,arh),4.96(s,2h,och2c=o),3.95(s,3h,och3),3.78(s,3h,arch3),3.07(s,3h,nch3),2.88(s,3h,nch3)。
实施例14
2-(4-((2,4-二甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二甲基乙酰胺(i-g′)的合成;
按照实施例8相同的方法制备,将原料苯胺替换为2,4-二甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-g′)0.148g,收率58%;m.p.:117.2~119.6℃;ir(v,cm-1):3366.56,2977.61,2944.60,1652.08,1474.07,1171.64,1035.23,806.79;1h-nmr(400mhz,dmso-d6):δ9.24(s,1h,nh),8.26(s,1h,c=nh),7.72(s,1h,arh),7.18(d,j=9.1hz,2h,arh),7.14(d,j=4.6hz,1h,arh),7.06(d,j=7.7hz,1h,arh),4.92(s,2h,och2c=o),3.94(s,3h,och3),3.05(s,3h,nch3),2.88(s,3h,nch3),2.31(s,3h,arch3),2.12(s,3h,arch3)。
实施例15
2-(7-甲氧基-4-(苯胺基)喹唑啉-6-基)氧二乙基乙酰胺(i-a″)的合成;
按照实施例1相同的方法制备到中间体4,将二乙胺z(1.12g,15.31mmol)、三乙胺(3.10g,30.63mmol)和二氯甲烷(10ml)混合于50ml单口瓶中,在-5℃冰浴和氮气保护条件下搅拌,缓慢加入氯乙酰氯(1.75g,15.47mmol)于上述液体中。1h后,将反应转移到室温,并在氮气保护下反应12h。tlc监测反应(二氯甲烷/乙酸乙酯=35/1),反应完全后,用饱和氯化铵水溶液(30ml)洗2次(15ml×2)。有机相再次用去离子水(15ml)清洗,无水硫酸镁干燥,过滤,浓缩得黄色透明液体2-氯-n,n-二乙基乙酰胺6z1.98g,收率86.42%。将侧链中间体密封,于阴凉干燥处保存留以备用。
将中间体4a(0.15g,0.50mmol)、2-氯-n,n-二乙基乙酰胺6z(0.112g,0.75mmol)和碳酸钾(0.138g,1.00mmol)混合溶于30ml干燥的n,n-二甲基甲酰胺中,在80℃下搅拌3h,tlc监测(二氯甲烷/甲醇=15/1)。反应毕,将混合物冷却至室温,然后缓慢加入冷水并搅拌,直至析出大量絮状固体。减压抽滤,干燥得黄色粉状固体2-(7-甲氧基-4-(苯胺基)喹唑啉-6-基)氧二乙基乙酰胺(i-a″)0.153g,收率65%;m.p.:182.0~184.5℃;ir(v,cm-1):3311.17,2973.88,2931.95,1636.17,1471.64,1392.10,1240.02,841.50;1h-nmr(600mhz,dmso-d6):δ9.39(s,1h,nh),8.46(s,1h,c=nh),7.82(s,1h,arh),7.78(d,j=7.9hz,2h,arh),7.39(t,j=7.8hz,2h,arh),7.11(d,j=7.3hz,1h,arh),4.93(s,2h,och2c=o),3.95(s,3h,och3),3.39(d,j=7.2hz,2h,ch2),3.32(d,j=7.0hz,2h,ch2),1.21(t,j=7.0hz,3h,ch3),1.06(t,j=7.0hz,3h,ch3)。
实施例16
2-(4-((2-氟苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二乙基乙酰胺(i-b″)的合成;
按照实施例15相同的方法制备,将原料苯胺替换为邻氟苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-b″)0.155g,收率63%;m.p.:184.8~185.2℃;ir(v,cm-1):3287.09,2974.94,2932.69,1633.98,1498.13,1426.48,1251.58,1202.61,842.90;1h-nmr(600mhz,dmso-d6):δ9.39(s,1h,nh),8.36(s,1h,c=nh),7.78(s,1h,arh),7.56(d,arh,j=7.4hz,1h,arh),7.31(d,j=7.8hz,2h,arh),7.25(d,j=6.6hz,1h,arh),7.22(s,1h,arh),4.91(s,2h,och2c=o),3.95(s,3h,och3),3.32(dd,j=13.9,6.9hz,4h,ch2),1.20(t,j=6.9hz,3h,ch3),1.06(t,j=6.9hz,3h,ch3)。
实施例17
2-(4-((4-氟-2-甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二乙基乙酰胺(i-c″)的合成;
按照实施例15相同的方法制备,将原料苯胺替换为4-氟-2-甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-c″)0.159g,收率73%;m.p.:117.4~118.6℃;ir(v,cm-1):3323.76,2976.18,2934.39,1621.82,1472.16,1389.40,1201.57,846.24;1h-nmr(600mhz,dmso-d6):δ9.22(s,1h,nh),8.28(s,1h,c=nh),7.76(s,1h,arh),7.34(dd,j=8.3,5.8hz,1h,arh),7.18(d,j=4.8hz,2h,arh),7.09(t,j=8.3hz,1h,arh),4.90(s,2h,och2c=o),3.94(s,3h,och3),3.38(d,j=7.0hz,2h,ch2),3.31(d,j=7.1hz,2h,ch2),2.17(s,3h,arch3),1.20(t,j=6.9hz,3h,ch3),1.06(t,j=7.0hz,3h,ch3)。
实施例18
2-(4-((4-甲氧基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二乙基乙酰胺(i-d″)的合成;
按照实施例15相同的方法制备,将原料苯胺替换为对甲氧基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-d″)0.161g,收率63%;m.p.:96.7~97.1℃;ir(v,cm-1):3300.16,3001.73,2936.22,2836.60,1696.72,1530.58,1470.63,1300.71,823.45;1h-nmr(600mhz,dmso-d6):δ9.38(s,1h,nh),8.50(s,1h,c=nh),7.83(s,1h,arh),7.50(s,1h,arh),7.40(s,1h,arh),7.28(s,1h,arh),7.23(s,1h,arh),6.71(s,1h,arh),4.94(s,2h,och2c=o),3.96(s,3h,och3),3.79(s,3h,arch3),3.40(d,j=6.6hz,2h,ch2),3.32(d,j=6.7hz,2h,ch2),1.22(t,j=5.8hz,3h,ch3),1.06(t,j=6.2hz,3h,ch3)。
实施例19
2-(4-((3-甲氧基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二乙基乙酰胺(i-e″)的合成;
按照实施例15相同的方法制备,将原料苯胺替换为间甲氧基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-e″)0.167g,收率63%;m.p.:174.6~175.8℃;ir(v,cm-1):3305.53,3008.96,2968.89,2938.31,1671.99,1433.66,1385.54,1222.95,837.74;1h-nmr(600mhz,dmso-d6):δ9.38(s,1h,nh),8.50(s,1h,c=nh),7.83(s,1h,arh),7.50(s,1h,arh),7.40(d,j=7.6hz,1h,arh),7.30(t,j=7.8hz,1h,arh),7.23(s,1h,arh),6.70(d,j=7.7hz,1h,arh),4.94(s,2h,och2c=o),3.96(s,3h,och3),3.79(s,3h,och3),3.40(d,j=6.6hz,2h,ch2),3.32(d,j=6.7hz,2h,ch2),1.22(t,j=5.8hz,3h,ch3),1.06(t,j=6.2hz,3h,ch3)。
实施例20
2-(4-((4-甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二乙基乙酰胺(i-f″)的合成;
按照实施例15相同的方法制备,将原料苯胺替换为对甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-f″)0.155g,收率67%;m.p.:106.9~108.2℃;ir(v,cm-1):3300.86,2972.50,2934.75,1618.38,1509.90,1424.17,1236.34,1201.26,844.89;1h-nmr(600mhz,dmso-d6):δ9.33(s,1h,nh),8.44(s,1h,c=nh),7.81(s,1h),7.65(d,j=8.2hz,2h),7.20(d,j=7.5hz,3h),4.92(s,och2c=o,2h),3.95(s,3h,och3),3.39(d,j=7.0hz,2h,ch2),3.32(d,j=7.0hz,2h,ch2),2.31(s,3h,arch3),1.22(t,j=6.9hz,3h,ch3),1.07(t,j=6.9hz,3h,ch3)。
实施例21
2-(4-((2,4-二甲基苯基)氨基)-7-甲氧基喹唑啉-6-基)-氧二乙基乙酰胺(i-g″)的合成;
按照实施例15相同的方法制备,将原料苯胺替换为2,4-二甲基苯胺,经氯代,胺化,酯的水解,酰胺化,烷基化5步反应合成目标化合物(i-g″)0.138g,收率58%;m.p.:187.3~189.1℃;ir(v,cm-1):3201.16,2975.54,2932.13,1658.20,1500.63,1423.28,1205.30,852.75;1h-nmr(400mhz,dmso-d6):δ9.19(s,1h,nh),8.27(s,1h,c=nh),7.77(s,1h,arh),7.21–7.05(m,4h,arh),4.90(s,2h,och2c=o),3.94(s,3h,och3),3.38(d,2h,ch2,j=7.7hz),3.32(d,2h,ch2,j=6.8hz),2.31(s,3h,arch3),2.12(s,3h,arch3),1.21(s,3h,ch3),1.06(s,3h,ch3)。
实施例22
片剂制备方法如下:
工艺:将活性成分辅料分别过100目筛,称取处方量的主药和辅料(一半羧甲基淀粉钠)充分混合,加入聚乙烯吡咯烷酮水溶液适量制软材,过24目筛,制得湿颗粒于50℃烘箱中干燥约2小时,将剩余羧甲基淀粉钠和硬脂酸镁与颗粒混合均匀,整粒,测定中间体含量,用φ8mm浅冲压片。
实施例23
注射液的制备
工艺:取注射用水30ml,称取处方量的柠檬酸、磷酸二氢钠搅拌使溶解,加入样品搅拌溶解,用0.1mol/l的盐酸或氢氧化钠调ph值为5.0,加入0.1%的活性炭吸附30分钟。用0.22μm精滤。按每安瓿5毫升灌装,108℃高温灭菌60分钟即得注射液。
实施例24
化合物i的体外抗肿瘤活性试验。
(1)材料;
细胞株:人胃癌细胞(mkn45)、人肺腺癌细胞(a549)、人肺癌细胞(h1975)。
试剂:mtt,amresco公司;dmem、dmem/f12培养基,gibco公司;小牛血清,兰州民海生物;胰蛋白酶,amresco公司。
仪器:超净工作台,苏州净化设备厂;co2培养箱,thermo公司,型号:heracell150;倒置显微镜,carlzeiss公司,型号:axiovert200;酶联免疫检测仪,tecan公司,型号:sunrise。
(2)方法;
细胞培养:细胞接种在含10%小牛血清,100iu/ml青霉素g钠盐及100ug/ml硫酸链霉素的dmem或dmem/f12完全培养液中,置37℃、100%相对湿度、含5%co2的培养箱中,传代3次后备用。
mtt比色法检测:取对数生长期的细胞,经0.25%胰蛋白酶消化后(悬浮细胞无须消化),悬浮于含10%小牛血清的培养液中,用玻璃滴管轻轻吹打成单细胞悬液,显微镜下用血细胞记数板记数活细胞。96孔培养板每孔接种细胞悬液90μl(细胞浓度为3~6×104个/ml),置培养箱24h后,每孔加10μl药液。另外,每个浓度设阴性对照(等浓度dmso)及空白本底(不加细胞),各组均设6个复孔。每孔加入100μldmso,置微量振荡器震荡5min以使结晶完全溶解,于酶标仪492nm单波长比色,测定od值,试验结果见表1。
抑制率(%)=[1-(实验组od均值-空白组od均值)/(对照组od均值-空白组od均值)]×100%。bliss法计算受试化合物ic50值。
(3)结果
表1对体外培养细胞生长的ic50(μmol/l)
从表1可以看出,相对吉非替尼,本发明所提供的喹唑啉衍生物大部分具有良好的药理活性。其中,喹唑啉衍生物i-b、i-b″、i-c″和i-d″对h1975细胞生长的抑制活性ic50值分别为0.720、0.842、0.478和0.652μmol/l,优于阳性对照药吉非替尼的6.925μmol/l;喹唑啉衍生物i-b′、i-g′和i-f″对mkn45细胞生长的抑制活性ic50值分别为0.376、0.532和0.324μmol/l,优于阳性对照药吉非替尼的3.281μmol/l。因此,所设计的喹唑啉衍生物具有更好的抑制活性。
唑啉衍生物i-b′对体外培养mkn45和h1975细胞生长的抑制活性ic50值分别为0.376μmol/l和18.411μmol/l;唑啉衍生物i-d″对体外培养mkn45和h1975细胞生长的抑制活性ic50值分别为7.548μmol/l和0.6521μmol/l。由此可见,所设计的喹唑啉衍生物具有更好的选择性,化合物i-b′有利于对mkn45细胞的抑制,而化合物i-d″有利于对耐药细胞株h1975的抑制。本发明所提供的部分喹唑啉衍生物具有良好的选择性。
- 还没有人留言评论。精彩留言会获得点赞!