乙烯基环丙基甲酸乙酯类化合物的制备方法与流程
1.本发明涉及一种乙烯基环丙基甲酸乙酯类化合物的制备方法。
背景技术:
2.(1r,2s)-1-氨基-2-乙烯基环丙基甲酸乙酯(结构式:)作为医药中间体应用比较广泛。
3.现有技术中该中间体的制备方法中,可利用手性诱导的相转移催化剂(例如等)通过关环直接得到80-90%手性纯度的产物,但以此类相转移催化剂进行关环反应时影响因素较多,尤其是在扩大反应规模时重复性较差,手性纯度不稳定。
4.利用此类相转移催化剂进行关环反应时,主要存在以下技术问题:影响手性纯度的因素较多,重复性较差。反应规模放大后反应速度非常缓慢(》3天),同时产物的手性纯度显著低于小规模反应。另外,需要进一步通过超临界流体色谱对关环反应后的产物进行手性分离,分离效率低,成本高,不适宜推广放大。以上技术问题亟待解决。
技术实现要素:
5.本发明所要解决的技术问题是为了克服现有制备工艺难以放大反应规模,且产物手性纯度较低,分离效率低的缺陷,而提供了一种乙烯基环丙基甲酸乙酯类化合物的制备方法,本发明方法催化剂效率高,易于放大,可重现性高,在大规模生产中显著降低成本。
6.本发明主要是通过以下技术方案解决上述技术问题的。
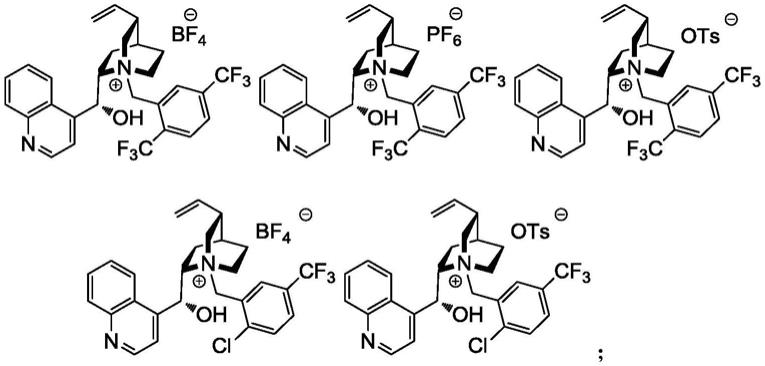
7.本发明提供了一种乙烯基环丙基甲酸乙酯类化合物的制备方法,其包括如下步骤:溶剂中,在如式a所示的化合物和碱的存在下,如式i所示的化合物与如式ii所示的化合物进行关环反应,得如式iii-1所示的化合物,即可;
8.9.式a中,r为cf3或cl,x为bf4、pf6或ots。
10.式a中,较佳地,r为cf3。
11.式a中,较佳地,x为bf4或pf6;更佳地,x为bf4。
12.本发明中,较佳地,所述的如式a所示的化合物选自如下任一化合物:
[0013][0014]
所述的关环反应中,所述的溶剂可为本领域此类反应的常用溶剂,例如苯类溶剂,再例如甲苯。
[0015]
所述的关环反应中,所述的溶剂的用量可为本领域此类反应的常规用量,较佳地,所述的溶剂与所述的如式i所示的化合物的体积质量比为10~20ml/g,例如12ml/g、15ml/g或18ml/g。
[0016]
所述的关环反应中,所述的如式a所示的化合物的用量可为本领域此类反应催化剂的常规用量,较佳地,所述的如式a所示的化合物与所述的如式i所示的化合物的摩尔比为(0.015~0.05):1,例如0.017:1、0.02:1、0.025:1、0.03:1、0.033:1、0.04:1或0.05:1。
[0017]
所述的关环反应中,所述的碱可为本领域此类反应常用的碱,例如naoh。所述的碱可以固体或水溶液的形式加入,例如以质量分数为30~50%的碱的水溶液的形式加入。
[0018]
所述的关环反应中,所述的碱用量可为本领域此类反应的常规用量,较佳地,所述的碱与所述的如式i所示的化合物的摩尔比为(5~15):1,例如6:1、10:1或12:1。
[0019]
所述的关环反应中,所述的如式i所示的化合物与所述的如式ii所示的化合物的摩尔比可为本领域此类反应的常规比例,较佳地,所述的如式i所示的化合物与所述的如式ii所示的化合物的摩尔比为1:(0.8~1.5),例如1:1、1:1.3或1.2:1。
[0020]
所述的关环反应中,所述的关环反应的温度可为本领域此类反应中常规的温度,本发明中较佳地为0℃~5℃,例如0℃。
[0021]
所述的关环反应中,所述的关环反应的进程可采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行检测,一般以所述的如式i或ii所示的化合物消失或不再反应时作为反应终点。所述的关环反应的时间可为5~20小时,例如5h、16h、18h或20h。
[0022]
本发明中,所述的关环反应还可包括如下后处理步骤:静置,分相,萃取,调节ph至8。
[0023]
本发明还提供了一种如式iv-1所示的化合物的制备方法,其包括如下步骤:
[0024]
(1)溶剂中,在如式a所示的化合物和碱的存在下,如式i所示的化合物与如式ii所示的化合物进行关环反应,得如式iii-1所示的化合物;
[0025][0026]
(2)溶剂中,如式iii-1所示的化合物与boc2o进行氨基保护反应,得如式iv-1所示的化合物,即可;
[0027][0028]
其中,r和x的定义如前所述,步骤(1)的具体反应条件和操作如前所述的乙烯基环丙基甲酸乙酯类化合物的制备方法所述。
[0029]
步骤(2)中,所述的溶剂可为本领域此类反应的常用溶剂,例如醚类溶剂,再例如甲基叔丁基醚(mtbe)。
[0030]
步骤(2)中,所述的溶剂的用量可为本领域此类反应的常规用量,较佳地,所述的溶剂与所述的如式iii-1所示的化合物的体积质量比为5~20ml/g。
[0031]
步骤(2)中,boc2o用量可为本领域此类反应的常规用量,较佳地,所述的如式iii-1所示的化合物与boc2o的摩尔比为1:(1~1.5),例如1:1.1。
[0032]
步骤(2)中,所述的氨基保护反应的温度可为本领域此类反应中常规的温度,本发明中较佳地为20℃~30℃,例如25℃。
[0033]
步骤(2)中,所述的氨基保护反应的进程可采用本领域中的常规监测方法(例如tlc、hplc或nmr)进行检测,一般以所述的如式iii-1所示的化合物消失或不再反应时作为反应终点。所述的关环反应的时间可为6~10小时,例如8h。
[0034]
步骤(2)中,所述的氨基保护反应还可包括如下后处理步骤:静置,分相,浓缩。
[0035]
步骤(2)中,所述的氨基保护反应的反应液中可包含杂质,例如包含如式iv-2所示的化合物如式iv-2所示的化合物的质量百分比可为25%以下,如式iv-2所示的化合物的质量百分比还可为22%以下,所述的质量百分比指如式iv-2所示的化合物的质量占“如式iv-1所示的化合物和式iv-2所示的化合物的混合物”总质量的百分比。
[0036]
所述的杂质如式iv-2所示的化合物可通过本领域常规的方法
除去,较佳地,步骤(2)中,所述的氨基保护反应还可包括如下手性纯化步骤:在酶的作用下,所述的氨基保护反应后的粗产物发生选择性水解反应。
[0037]
所述的手性纯化步骤中,所述的氨基保护反应后的粗产物通常指步骤(2)中经所述的氨基保护反应后,经所述后处理得到的粗产物。
[0038]
所述的手性纯化步骤中,如式iv-2所示的化合物在酶的作用下水解为如式iv-3所示的化合物,由此减少粗产物中如式iv-2所示的化合物的含量,提高目标产物的手性纯度。
[0039][0040]
所述的手性纯化步骤中,可在溶剂中进行所述的选择性水解反应,所述的溶剂可为本领域此类反应的常用溶剂,例如dmso。所述的溶剂的用量可为本领域此类反应的常规用量,较佳地,所述的溶剂与所述的氨基保护反应后的产物的体积质量比的用量范围为1~10ml/g(4ml/g)。
[0041]
所述的选择性水解反应中,所述的酶可为本领域此类反应常用的酶,例如蛋白水解酶(例如蛋白水解酶2.4l fg alcalase)。
[0042]
所述的选择性水解反应中,所述的酶以含酶的缓冲液形式加入至体系中。较佳地,所述的含酶的缓冲液中,所述的缓冲液包含磷酸二氢钾和三水合磷酸氢二钾(例如含1.44g/l的磷酸二氢钾和43.22g/l的三水合磷酸氢二钾)。较佳地,所述的含酶的缓冲液中,所述的酶与所述的缓冲液的体积质量比为3~5ml/g(例如3.5ml/g)。
[0043]
所述的选择性水解反应中,所述的酶的用量可为本领域此类反应的常规用量,较佳地,所述的酶与所述的氨基保护反应后的粗产物的质量比为(2~4):1(例如3:1)。
[0044]
本发明还提供了一种如式a所示的化合物:
[0045][0046]
其中,r为cf3或cl,x为bf4、pf6或ots。
[0047]
式a中,较佳地,r为cf3。
[0048]
式a中,较佳地,x为bf4或pf6;更佳地,x为bf4。
[0049]
本发明中,较佳地,所述的如式a所示的化合物选自如下任一化合物:
[0050][0051]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0052]
本发明所用试剂和原料均市售可得。
[0053]
本发明的积极进步效果在于:本发明方法纯度高,立体选择性好,且反应稳定,可重现性高。
[0054]
本发明方法催化剂效率高,易于放大,在大规模生产中显著降低成本。
具体实施方式
[0055]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0056]
以下实施例中,使用磁力搅拌(300-350rpm)。
[0057]
nmr结果:在bruker 400mhz光谱仪上记录核磁共振(nmr)光谱,dmso作为氘代溶剂;
[0058]
hplc纯度:色谱柱:waters xbridge c18(4.6
×
150mm,3.5μm);流动相:乙腈/水(v/v=1:4),流速:1ml/分钟,检测器:254nm;
[0059]
手性纯度:手性色谱柱:chiralcel oj-h(150mm
×
4.6mm
×
5μm);流动相:正庚烷/乙醇(v/v=9:1),流速:1ml/分钟,检测器:254nm。
[0060]
实施例1
[0061][0062]
(1)向反应器中加入化合物a(100.0g,0.34mol)、化合物b(125.0g,0.41mol)和1300g甲苯,加热至110℃,搅拌反应4小时,监控反应进程。将反应器中的反应液过滤,并使用甲苯(20g)洗涤,得滤饼,即化合物a-0。
[0063]
(2)将得到的滤饼转移至反应器中,并加入nabf4(112g,1.02mol)和1300g ch3cn,在25℃下搅拌24小时,浓缩后加入1300g水,并用1300g dcm萃取,浓缩后取样进行分析,得化合物a-1(摩尔收率85%)。
[0064]1h nmr(400mhz,dmso-d6)δppm 1.04-1.42(m,1h)1.69-1.84(m,1h)2.00-2.19(m,3h)2.68(br s,1h)3.14-3.28(m,1h)3.38-3.62(m,1h)3.93-4.07(m,2h)4.30-4.41(m,1h)4.96(d,j=10.54hz,1h)5.17-5.33(m,2h)5.52(d,j=13.55hz,1h)5.62-5.76(m,1h)6.58(br s,1h)6.90(br s,1h)7.75-7.90(m,3h)8.14(d,j=8.07hz,1h)8.23-8.31(m,3h)8.48(s,1h)9.02(d,j=4.53hz,1h)。
[0065]
使用ic离子色谱仪,以nabf4作对照标准品,将化合物a-1与nabf4在相同的条件下经离子色谱仪分析,色谱条件如表1所示,
[0066]
表1
[0067]
[0068][0069]
结果显示,化合物a-1与nabf4的出峰在同一保留时间(4min),且化合物a-1的色谱图中无其他明显特征峰。
[0070]
实施例2
[0071][0072]
向反应器中加入化合物a(100.0g,0.34mol)、化合物b(125.0g,0.41mol)和1300g甲苯,加热至110℃,搅拌反应4小时,监控反应进程。将反应器中的反应液过滤,并使用甲苯(20g)洗涤,得滤饼,即化合物a-0。
[0073]
将得到的滤饼转移至反应器中,并加入kpf6(188g,1.02mol)和1300g ch3cn,在25℃下搅拌24小时,浓缩后加入1300g水,并用1300g dcm萃取,浓缩后取样进行分析,得化合物a-2(摩尔收率83%)。
[0074]1h nmr(400mhz,dmso-d6)δppm 1.12-1.32(m,1h)1.68-1.87(m,1h)1.98-2.20(m,3h)2.56-2.73(m,1h)3.16-3.27(m,1h)3.53(br t,j=11.17hz,1h)3.92-4.09(m,2h)4.28-4.44(m,1h)4.96(d,j=10.54hz,1h)5.15-5.36(m,2h)5.53(br d,j=13.55hz,1h)5.60-5.76(m,1h)6.58(br s,1h)6.91(d,j=2.76hz,1h)7.74-7.91(m,3h)8.14(d,j=8.28hz,1h)8.23-8.32(m,3h)8.49(s,1h)9.02(d,j=4.62hz,1h)。
[0075]
使用ic离子色谱仪,以kpf6作对照标准品,将化合物a-2与kpf6在相同的条件下经
离子色谱仪分析,色谱条件同表1,结果显示化合物a-2与kpf6的出峰在同一保留时间(12min),且化合物a-2的色谱图中无其他明显特征峰。
[0076]
实施例3
[0077][0078]
参照实施例1中化合物a-1的合成方法,分别使用甲磺酸钠、对甲苯磺酸钠和全氟丁基磺酸钠替换nabf4与化合物a-0进行盐交换,分别合成了化合物a-3、化合物a-4和化合物a-5。
[0079]
化合物a-4:1h nmr(400mhz,dmso-d6)δppm 1.11-1.36(m,2h)1.75(br d,j=5.25hz,1h)1.98-2.19(m,3h)2.22-2.35(m,3h)2.67(br dd,j=3.69,1.94hz,1h)3.16-3.33(m,2h)3.51(t,j=11.19hz,1h)3.92-4.05(m,2h)4.33(br s,1h)4.95(d,j=10.63hz,1h)5.17-5.31(m,2h)5.51(d,j=13.76hz,1h)5.66(ddd,j=17.26,10.76,6.25hz,1h)6.56(br s,1h)6.89(d,j=2.75hz,1h)7.10(d,j=7.57hz,2h)7.47(d,j=7.65hz,2h)7.70-7.89(m,3h)8.13(d,j=8.12hz,1h)8.22-8.29(m,3h)8.47(s,1h)9.01(d,j=4.58hz,1h)。
[0080]
使用ic离子色谱仪,以naots作对照标准品,将化合物a-4与naots在相同的条件下经离子色谱仪分析,色谱条件同表1,结果显示化合物a-4与naots的出峰在同一保留时间(8.7min),且化合物a-4的色谱图中无其他明显特征峰。
[0081]
化合物a-5:h nmr(400mhz,dmso-d6)δppm 1.22(br t,j=10.04hz,1h)1.68-1.86(m,1h)1.99-2.19(m,3h)2.68(br s,1h)3.16-3.34(m,1h)3.39-3.57(m,1h)3.93-4.06(m,2h)4.30-4.40(m,1h)4.96(d,j=10.54hz,1h)5.16-5.33(m,2h)5.52(d,j=13.55hz,1h)5.67(ddd,j=17.19,10.67,6.27hz,1h)6.58(br s,1h)6.89(d,j=2.76hz,1h)7.75-7.90(m,3h)8.14(d,j=8.03hz,1h)8.22-8.31(m,3h)8.48(s,1h)9.01(d,j=4.61hz,1h)。
[0082]
使用ic离子色谱仪,全氟丁基磺酸钾作对照标准品,将化合物a-5与全氟丁基磺酸
钾在相同条件下经离子色谱仪分析,色谱条件如表2所示,
[0083]
表2
[0084][0085][0086]
结果显示化合物a-5与全氟丁基磺酸钾的出峰在同一保留时间(16.8min),且化合物a-5的色谱图中无其他明显特征峰。
[0087]
实施例4
[0088][0089]
化合物a-6的合成参照实施例1中化合物a-0的合成方法,用2-溴甲基-1-氯-4-(三氟甲基)苯替代2-溴甲基-1,4-双(三氟甲基)苯,得化合物a-6。
[0090]
实施例5
[0091][0092]
参照实施例3的方法,分别使用nabf4、对甲苯磺酸钠和全氟丁基磺酸钠与化合物a-6进行盐交换,分别合成了化合物a-7、化合物a-8和化合物a-9。
[0093]
化合物a-7:1h nmr(400mhz,dmso-d6)δppm 1.14-1.36(m,1h)1.80(br s,1h)1.97-2.20(m,3h)2.68(br s,1h)3.19-3.34(m,1h)3.41-3.61(m,1h)3.88-4.06(m,2h)4.47(br s,1h)4.97(d,j=10.54hz,1h)5.11-5.25(m,2h)5.45(br d,j=13.05hz,1h)5.60-5.77(m,1h)6.60(br s,1h)6.91(d,j=3.26hz,1h)7.75-7.90(m,3h)7.96-8.06(m,2h)8.13(d,j=8.28hz,1h)8.30(d,j=8.28hz,1h)8.40(s,1h)9.01(d,j=4.64hz,1h)。
[0094]
化合物a-8:1h nmr(400mhz,dmso-d6)δppm 1.12-1.35(m,1h)1.79(br s,1h)1.97-2.19(m,3h)2.22-2.32(m,2h)2.68(br s,1h)3.19-3.35(m,1h)3.50(br t,j=11.29hz,1h)3.87-4.07(m,2h)4.46(br s,1h)4.96(d,j=10.54hz,1h)5.11-5.24(m,2h)5.44(br d,j=13.05hz,1h)5.68(ddd,j=17.19,10.67,6.27hz,1h)6.59(br s,1h)6.92(d,j=3.51hz,1h)7.06-7.20(m,2h)7.47(d,j=8.03hz,1h)7.70-7.90(m,3h)7.95-8.05(m,2h)8.13(d,j=8.03hz,1h)8.30(d,j=8.28hz,1h)8.39(s,1h)9.00(d,j=4.56hz,1h)。
[0095]
化合物a-9:1h nmr(400mhz,dmso-d6)δppm 1.25(br t,j=11.04hz,1h)1.80(br s,1h)1.96-2.20(m,3h)2.55-2.73(m,1h)3.18-3.34(m,1h)3.39-3.60(m,1h)3.85-4.05(m,2h)4.45(br s,1h)4.97(d,j=10.54hz,1h)5.07-5.25(m,2h)5.43(br d,j=13.05hz,1h)5.68(ddd,j=17.19,10.67,6.27hz,1h)6.59(br s,1h)6.88(d,j=3.26hz,1h)7.74-7.91(m,3h)7.96-8.06(m,2h)8.13(d,j=8.28hz,1h)8.28(d,j=8.28hz,1h)8.37(s,1h)9.01(d,j=4.52hz,1h)。
[0096]
化合物a-7、化合物a-8和化合物a-9的阴离子分析结果与化合物a-1、化合物a-4和化合物a-5相同。
[0097]
实施例6
[0098][0099]
化合物a-10的合成参照实施例1中化合物a-0的合成方法,用9-氯甲基蒽替代2-溴甲基-1,4-双(三氟甲基)苯,得化合物a-10。
[0100]
实施例7
[0101][0102]
参照实施例3的方法,分别使用nabf4与化合物a-10进行盐交换,分别合成了化合物a-11。
[0103]
实施例8应用手性诱导的相转移催化剂进行关环反应
[0104][0105]
向反应器中加入化合物i、化合物ii、催化剂(如表3所示,化合物a-0~化合物a-11)和甲苯,冷却至0℃,在0℃下搅拌1h。在0℃下加入质量分数为50%的naoh水溶液,在0℃下继续搅拌进行反应,反应过程中取样进行分析,得化合物iii,其中,化合物iii为化合物iii-1和化合物iii-2的混合物。
[0106]
表3
[0107][0108][0109]
反应条件及结果如表4所示:
[0110]
表4
[0111][0112][0113]
实施例9催化剂用量
[0114][0115]
向反应器中加入化合物i(1.1g,1.2eq)、化合物ii(1.0g,1.0eq)、化合物a-2(用量如表5所示)和20ml甲苯,冷却至0℃,在0℃下搅拌1h。在0℃下加入质量分数为50%的naoh水溶液(4.5g,12eq),在0℃下继续搅拌进行反应,反应过程中取样进行分析,得化合物iii,其中,化合物iii为化合物iii-1和化合物iii-2的混合物。
[0116]
结果如表5所示。
[0117]
表5
[0118][0119]
由以上结果可知,本发明催化剂可以由5mol%或以上降低到2mol%以下而不明显影响反应速度和手性选择性,使催化剂效率得到提高,在大规模生产中可以显著降低成本。
[0120]
实施例10反应规模
[0121][0122]
向反应器中加入化合物i(50g,1.2eq)、化合物ii(46.6g,1.0eq)、化合物a-1
(3.98g,0.03eq)和600ml甲苯,冷却至0℃,在0℃下搅拌1h。在0℃下加入质量分数为50%的naoh水溶液(225g),在0℃下继续搅拌进行反应18h,反应过程中取样进行分析,得化合物iii,其中,化合物iii为化合物iii-1和化合物iii-2的混合物。
[0123]
反应18h后得化合物iii 54g收率84.9%,产物的hplc纯度为84.1%,手性纯度(化合物iii-1/化合物iii-2)为76.6/23.4。
[0124]
实施例11
[0125][0126]
向反应器中加入化合物i(100.0g,0.52mol)、化合物ii(89.0g,0.42mol)、化合物a-1(9.4g,15.6mmol)和1300g甲苯,冷却至0℃,在0℃下搅拌1h。在0℃下加入416g质量分数为50%的naoh水溶液,在0℃下继续搅拌16h,取样进行分析,产物的hplc纯度为85.6%,手性纯度(化合物iii-1/化合物iii-2)为77.3/22.7。
[0127]
静置1h后分相,取上层有机相,加入1000g 2m hcl水溶液,在25℃下搅拌4h。
[0128]
静置1h后分相,取下层水相,在25℃下使用10m naoh水溶液调节ph至8,加入boc2o(124.8g,0.57mol)和600g mtbe,在25℃下搅拌8h。
[0129]
静置1h后分相,取上层有机相,并用700g mtbe萃取水相,合并有机相后浓缩,取样分析,产物的hplc纯度为92.4%,手性纯度(化合物iii-1/化合物iii-2)为77.2/22.8,得产物,即化合物iv 85g,收率80.5%。
[0130]
实施例12酶催化手性选择性水解
[0131][0132]
将15.0g酶(诺维信蛋白水解酶2.4l fg alcalase)加入至52.5ml的缓冲液(缓冲液含1.44g/l的磷酸二氢钾和43.22g/l的三水合磷酸氢二钾)中,得含酶的缓冲液。
[0133]
取5.0g实施例11得到的产品加入至20ml的dmso,搅拌至溶解,加入前述含酶的缓冲液,调整ph至7.5。在45℃下搅拌,反应过程中取样进行分析,结果如表6所示。反应118h后,用50g 2-methf萃取水相两次,合并有机相后浓缩得产物,即化合物iv-1 3.7g,收率75%。
[0134]
表6
[0135]
反应时间(h)手性纯度(iv-1/iv-2)2489.447/10.5534694.760/5.247098.007/1.9939499.141/0.85911899.548/0.452
[0136]
对比例1反应规模
[0137][0138]
向反应器中加入化合物i(1.0eq)、化合物ii(1.3eq)、化合物a-0(0.05eq)和甲苯,冷却至0℃,在0℃下搅拌1h。在0℃下加入质量分数为50%的naoh水溶液(10eq),在0℃下继续搅拌进行反应,反应过程中取样进行分析,得化合物iii,其中,化合物iii为化合物iii-1和化合物iii-2的混合物。反应条件及结果如表7所示。
[0139]
表7
[0140][0141]
由以上结果可知,使用化合物a-0作为催化剂时,化合物i的反应规模增大至66g时,反应的速率显著低于小规模,使用0℃下搅拌40小时后,几乎没有产物生成,随后将温度升高至25℃并继续搅拌20h,hplc显示产品纯度仅为37.3%,随后在25℃下搅拌继续60h,最后得到hplc纯度为87.5%,手性纯度为71.3%的产品。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1