一种内标组合物及其在检测动物病原微生物中的应用的制作方法
本发明涉及动物病原微生物检测技术领域,尤其涉及一种内标组合物及其在检测动物病原微生物中的应用。
背景技术:
实时荧光定量pcr技术具有特异性强、灵敏度高、重复性好、定量准确、速度快、全封闭反应等优点,且克服常规pcr不能准确定量和易污染引起假阳性的问题,已被广泛应用于动物病原微生物的检测。以往在动物病原微生物的检测很少用到内标。然而,近些年来的研究表明,采用实时荧光pcr方法检测动物病原微生物时,常常会受到来自环境因素和样品中理化性质的干扰,特别是一些能够阻碍pcr反应的抑制物,会使检测结果呈假阴性。在实时荧光定量pcr检测中,假阴性一般是由于pcr扩增失败导致的,如提取过程中靶基因的丢失、提取试剂残留、标本中抑制物去除不彻底、扩增仪孔间温度差异等。现有技术中,避免假阴性最有效的措施是建立“内标”。研究表明,复杂的动物样品成分(组织、血液、唾液、粪便等),培养基成分以及残留的核酸提取试剂等抑制因子,可能会对实时荧光定量pcr反应产生抑制作用,影响检测结果的准确性。因此,在检测体系中添加用于监测核酸提取或指示反应抑制情况的内标则显得尤为重要。动物样品中病原微生物荧光定量pcr检测技术体系中的内标对于确保检测结果的可靠性具有重要的作用,一个可靠、有效的动物病原微生物检测方法必须包含有可以指示假阴性结果的内标,可有效监测核酸提取、扩增中出现的误差,从而避免假阴性结果。
制备内标的方法有很多,根据不同的实验目的和要求采用不同的制备方法。初期的内标是利用克隆技术将目的序列的pcr产物通过插入、删除或者替换等手段处理后制备的,制备方法较繁琐且制备的内标仅能用于单一病原微生物的检测。因此,需要开发出一种通用的异源性内标用于不同目的基因的动物病原微生物的检测,不仅可以用于单一病原微生物的检测,而且还可以用于多个病原微生物的检测。
增强型绿色萤光蛋白(enhancedgreenfluorescentprotein,egfp)是水母体内的绿色荧光蛋白(greenfluorescentprotein,gfp)基因经改造后的突变体,在蓝色450~490nm波长范围的光线激发下,显示绿色萤光,比通用的gfp荧光显示程度更强,检测灵敏度更高。作为良好的标记和报告基因,gfp基因应用范围非常广泛。目前gfp及其突变体egfp作为基因表达标记物或融合蛋白标记在动物、植物、微生物的研究中广泛应用。
技术实现要素:
为了克服现有技术中所存在的缺陷,本发明提供一种内标组合物及其在检测动物病原微生物中的应用,其选取egfp的一段保守基因序列,由于序列来源于发光水母基因,与genbank数据库中所收录的动物病原微生物基因组没有任何同源性,防止与目标基因同时检测时出现非特异性扩增,因此其可作为一种通用的异源性实时荧光定量pcr内标以用于不同目的基因的动物病原微生物的检测,将内标组合物应用于荧光定量pcr,以有效监测核酸提取、荧光pcr扩增中出现的误差,从而避免假阴性的结果。
为实现上述目的,本发明采用如下技术方案:
本发明的第一个方面是提供一种内标组合物,其包括含内标基因序列的质粒和用于扩增所述内标基因序列的引物探针组合;其中,所述内标基因序列为增强型绿色萤光蛋白(egfp)的保守基因序列。
为了进一步优化上述内标组合物,本发明采取的技术措施还包括:
进一步地,所述内标基因序列与动物病原微生物基因组没有任何同源性。
进一步地,所述内标基因序列如seqidno.1所示。上述内标基因序列为egfp的保守基因序列,长度约200bp。
进一步地,所述引物探针组合包括如seqidno.2和seqidno.3所示序列的第一内标引物对或如seqidno.2和seqidno.4所示序列的第二内标引物对;和/或,如seqidno.5所示序列的内标探针。
进一步地,所述内标探针在5’端标记荧光报告基团,在3’端标记荧光淬灭基团。其中,所述荧光报告基团选自fam、hex、vic、cy3、cy5、rox等,所述荧光淬灭基团选自bhq1、bhq2与bhq3、tamra等。可理解的是,上述荧光报告基团和荧光淬灭基团可为领域常规使用的任一合适的基团。
进一步地,所述内标探针上的荧光报告基团与待检测目的基因的探针上的荧光报告基团不同。
本发明的第二个方面是提供一种任一上述的内标组合物作为检测动物病原微生物的内标的应用。
为了进一步优化上述应用,本发明采取的技术措施还包括:
进一步地,所述第一内标引物对和内标探针用于检测单一动物病原微生物,所述第二内标引物对和内标探针用于检测多种动物病原微生物,优选2~4种。在一具体案例中,单一动物病原微生物为禽流感病毒,多种动物病原微生物包括猫流感病毒和猫疱疹病毒。
进一步地,在用于检测单一或多种动物病原微生物的内标探针采用的荧光报告基团和荧光淬灭基团均不同;其中,用于检测单一动物病原微生物的内标探针,其荧光报告基团为hex,荧光淬灭基团为bhq1;用于检测多种动物病原微生物的内标探针,其荧光报告基团为rox,荧光淬灭基团为bhq2。
可理解的是,上述内标组合物适用于任何适宜采用其作为内标的检测系统,并不仅仅局限于动物病原微生物的检测。
本发明的第三个方面是提供一种基于非诊断目的的采用任一上述的内标组合物检测动物病原微生物的实时荧光定量pcr方法,其包括以下步骤:
步骤1)将含内标基因序列的质粒添加到含样品的荧光pcr反应体系中;
步骤2)在所述反应体系中,采用第一内标引物对和内标探针或第二内标引物组合物和内标探针进行内标基因的扩增,同时采用引物和探针进行样品目的基因的扩增;
步骤3)对扩增结果进行分析。
为了进一步优化上述检测方法,本发明采取的技术措施还包括:
进一步地,所述步骤(1)中,含内标基因序列的质粒参与样品的核酸提取后添加到含荧光pcr反应体系中或者直接添加至含样品的荧光pcr反应体系中。
进一步地,当样品中含有目的基因时,内标基因与目的基因均有s型扩增曲线,检测结果为阳性;当样品中不含有目的基因时,只有内标基因有s型扩增曲线,检测结果为阴性;当无任何s型扩增曲线时,检测结果即为假阴性。
进一步地,所述假阴性是指荧光pcr反应受到了样品中存在的抑制剂(血液中的抗凝剂,口液中的酶类等)的影响而没有发生反应,或者由于操作人员的操作失误导致结果呈现阴性。
进一步地,在一具体案例中,所述荧光pcr反应体系中添加1.0×105copies/μl的内标。
进一步地,在一具体案例中,所述荧光pcr反应体系的扩增程序为42℃5min,95℃预变性10min;95℃15s,60℃30s,40个循环。
本发明的第四个方面是提供一种含有内标基因序列的质粒的构建方法,其包括以下步骤:
步骤a.选取内标基因序列:增强型绿色萤光蛋白的保守基因序列,序列如seqidno.1所示;根据所述内标基因序列设计如seqidno.2和seqidno.3所示序列的第一内标引物对或如seqidno.2和seqidno.4所示序列的第二内标引物对以及如seqidno.5所示序列的内标探针;
步骤b.将合成的含内标基因序列的dna片段与质粒载体连接,转化大肠杆菌,筛选大肠杆菌阳性克隆;
步骤c.利用所述第一内标引物对和内标探针或所述第二内标引物对和内标探针对所筛选的大肠杆菌阳性克隆进行荧光pcr检测验证,以构建含内标基因序列的质粒。
为了进一步优化上述构建方法,本发明采取的技术措施还包括:
进一步地,所述构建含内标序列的质粒具体包括:采用转基因方法将上述带有内标基因序列的载体转移到大肠杆菌中,然后涂布于选择性培养基平板上,经37℃培养12h,从选择性培养基平板上挑选单菌落(阳性克隆),接种于5ml含有氨苄青霉素的lb液体培养基中,置于37℃转速200r/min的摇床中培养8h,提取质粒,经pcr扩增后,确定获得转化子。
进一步地,所述载体,其转化的宿主细胞是大肠杆菌,其中包含有内标基因序列的dna扩增片段。
进一步地,所述选择性培养基平板,是指90mm选择性培养基平板,其中含有100mg/ml氨苄青霉素10μl,20%的iptg(200mg/ml)溶液7μl,2%的x-gal(20mg/ml)溶液40μl。
进一步地,所述步骤b中质粒载体连接具体包括:用ecorⅰ双酶切pgem-teasy克隆载体,将合成的含内标基因序列的dna片段与pgem-teasy载体在连接酶和连接缓冲液作用下16℃过夜。
进一步地,所述步骤c中还包括对荧光pcr检测所确定的大肠杆菌阳性克隆进一步的测序验证。
本发明的第五个方面是提供一种用于检测动物病原微生物的试剂盒,包括检测试剂和任一上述的内标组合物。
为了进一步优化上述试剂盒,本发明采取的技术措施包括:
进一步地,在一具体案例中,所述检测试剂包括pcr反应试剂,包括:10×buffer、mgcl2溶液(25mm)、dntpmixture、extaqdna聚合酶、反转录酶、目的基因上下游引物和探针、ddh2o。
上述内标基因、引物和探针的序列具体如下表所示:
表1-内标基因、引物和探针的序列
与现有技术相比,本发明采用上述技术方案,具有如下技术效果:
本发明根据egfp的广泛应用,选取其保守基因序列约200bp,采用分子生物信息学方法,设计3条引物(ic-f、ic-r1和ic-r2)和1条探针(ic-p,两种标记方式hex和rox),组合成两种内标:一种为ic-f/ic-r1和ic-p-hex;一种为ic-f/ic-r2和ic-p-rox,可用于核酸提取的监控,也可用于核酸扩增的监控;不仅可用于单一病原微生物的检测,也可用于多种(2~4种)病原微生物的检测,确保了检测的准确性和高效性,而且大大降低了检测成本,为动物病原微生物的荧光定量pcr检测的规范化和标准化提供了技术支撑。
本发明将所构建的内标添加到样品中进行核酸抽提或直接添加到荧光pcr反应体系中,可有效监测核酸提取和产物扩增出现的误差,避免假阴性的发生,从而提高荧光pcr检测的准确率,为动物病原微生物的检测提供有效可靠的技术手段。
附图说明
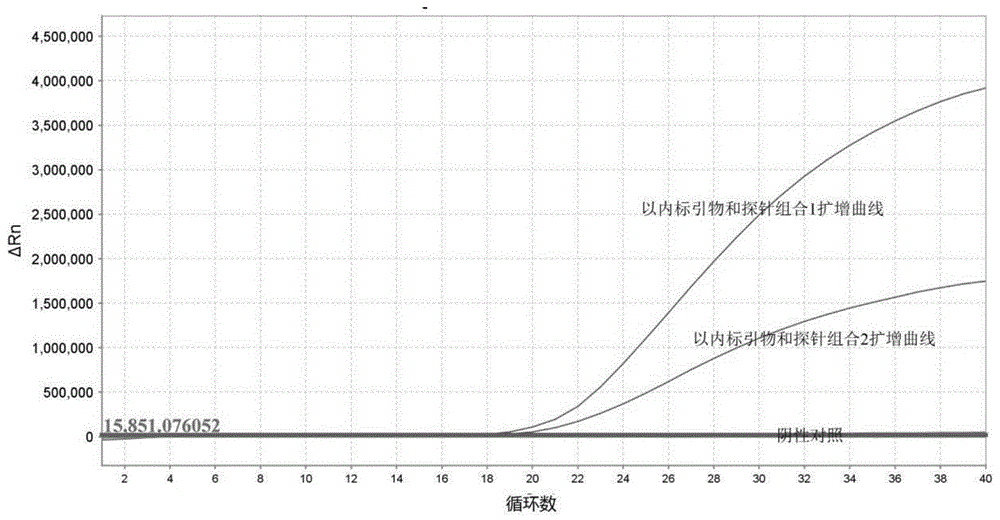
图1为本发明一实施例中大肠杆菌阳性克隆荧光pcr检测结果示意图;
图2为本发明一实施例中荧光pcr检测所确定的大肠杆菌阳性克隆进一步的测序验证的结果示意图;
图3为本发明一实施例中添加内标的不同浓度禽流感病毒质粒的荧光pcr检测验证结果示意图;
图4为本发明一实施例中添加内标的不同浓度猫流感病毒和猫疱疹病毒质粒的多重荧光pcr检测验证结果示意图。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。下列实施例中未注明具体条件的实验方法,按照本领域常规方法和条件,或按照商品说明书(或按照制造厂商的建议)选择。
实施例1-内标组合物的构建
本实施例为内标组合物的构建,其包括下述步骤:
1、内标序列的选取
选取egfp(accessionno.u55761)的一段保守基因序列(如上表1中的seqidno.1所示),长约200bp,序列来源于发光水母基因,与genbank数据库中所收录的动物病原微生物基因组没有任何同源性,防止与目标基因同时检测时出现非特异性扩增,因此可作为实时荧光定量pcr的内标。
2、内标引物和探针的设计
根据egfp基因序列seqidno.1,采用primer5.0和primerexpress3.0软件设计内标引物和探针,一条上游引物ic-f(如上表1中的seqidno.2所示)和两条下游引物[ic-r1(如上表1中的seqidno.3所示)和ic-r2(如上表1中的seqidno.4所示)],一条探针ic-p(如上表1中的seqidno.5所示)。上述探针ic-p采用两种荧光标记:一种为探针ic-p-hex,其荧光标记选择hex作为报告发光团,bhq1为淬灭基团;另一种为探针ic-p-rox,其荧光标记选择rox作为报告发光基团,bhq2为淬灭基团。选取内标探针时,应选取与目的基因探针上不同标记的荧光报告基团。选取引物(ic-f和ic-r1)和探针(ic-p-hex)组合1作为内标,扩增长为132bp,可在检测单一动物病原微生物时使用。选取引物(ic-f和ic-r2)和探针(ic-p-rox)组合2作为内标,扩增长为177bp,可在检测多种(2~4种)动物病原微生物时使用。
3、合成内标基因
将egfp基因序列seqidno.1委托生工生物工程(上海)上海股份有限公司合成。
4、连接
用ecorⅰ双酶切pgem-teasy克隆载体,将上述合成的含egfp基因序列seqidno.1的dna片段与pgem-teasy载体混合配制成体积为10μl的dna溶液,加入10μl的dna连接酶混合液(ligationmix),充分混合,在连接酶和连接缓冲液作用下16℃过夜。
5、转化大肠杆菌
用氯化钙法转化大肠杆菌dh5α,然后涂布于选择性培养基平板上,选择性培养基平板具体为:90mm培养皿,100mg/ml氨苄青霉素10μl,20%的iptg(200mg/ml)溶液7μl,2%的x-gal(20mg/ml)溶液40μl,37℃培养12h,从选择性平板上挑选白色菌落(即筛选阳性克隆),接种于5ml含有氨苄青霉素的lb液体培养基中,置于37℃转速200r/min的摇床中培养8h,提取质粒。
6、检测
利用内标特异性引物和探针(组合1和组合2)对上述提取的质粒进行荧光pcr检测,具体结果如图1所示,其中曲线1以内标引物和探针组合1扩增的结果;曲线2以内标引物和探针组合2扩增的结果;曲线3阴性对照,由上述结果可知内标特异性引物和探针可对内标基因序列进行特异性扩增。对荧光pcr检测所确定的大肠杆菌阳性克隆进一步的测序验证,其结果如图2所示,结果显示获得的含内标基因序列的质粒序列正确。
将上述内标特异性引物和探针(组合1或组合2)和构建的含内标基因序列的质粒形成内标组合物,即内标组合物包括:含内标基因序列的质粒、引物(ic-f和ic-r1)和探针(ic-p-hex);或者,含内标基因序列的质粒、引物(ic-f和ic-r2)和探针(ic-p-rox)。
实施例2-内标组合物的应用
本实施例为将实施例1构建的内标组合物作为内标应用于动物病原微生物的检测。
为了评价内标在单一或多重实时荧光pcr检测体系中的应用效果,用含有1.0×105copies/μl的内标添加到单一或多重实时荧光pcr反应体系中,对反应体系中靶基因dna的扩增的抑制程度、扩增曲线及效率进行评价。
1、在单一病原检测中的验证
(1)添加内标的实时荧光定量pcr反应体系的构建
反应总体积为25μl,10×buffer2.5μl,mgcl2溶液(25mm)1.5μl,dntpmixture(2.5mmol/l)1μl,extaqdna聚合酶(2.5u)0.5μl,反转录酶0.5μl,禽流感病毒上下游引物各1μl,禽流感病毒探针0.5μl,内标上下游引物各0.5μl,内标探针0.5μl,1.0×105copies/μl的内标1μl,模板dna5μl,补足ddh2o。扩增程序:42℃5min,95℃预变性10min;95℃15s,60℃30s,40个循环。
(2)添加内标的实时荧光定量pcr反应体系的评价
提取含禽流感病毒目标基因的质粒dna,经浓度测定进行一系列的10倍稀释后,以1.6×107、1.6×106、1.6×105、1.6×104拷贝/μl加入到添加含内标的禽流感病毒实时荧光pcr反应体系中,用以测定该pcr体系的扩增情况和内标的扩增情况,其扩增结果如图3所示。
结果表明,含禽流感病毒目标基因的质粒dna浓度在1.6×104拷贝/μl~1.6×107拷贝/μl时,内标的扩增均没有受到影响。虽然当dna的浓度在1.6×107拷贝/μl时,内标的扩增受到了一点抑制,但此时含禽流感病毒目标基因的质粒dna很容易被检测出,因此,在这种情况下内标的扩增并不重要,因为只有在内标和检测目标dna的扩增均受到抑制的情况下,才能说明pcr抑制物的存在,检测结果为假阴性,因而并不影响内标作为假阴性结果指示物的功能。同时,在添加了内标时,禽流感病毒的扩增曲线呈典型的s型,扩增效率很高,没有受到抑制,说明添加内标对实时荧光pcr扩增体系中禽流感病毒的扩增没有任何影响。
2、在多重病原检测中的验证
提取含猫流感病毒和猫疱疹病毒目标基因的质粒dna,经浓度测定进行一系列的10倍稀释后,以1.6×107、1.6×106、1.6×105、1.6×104拷贝/μl加入到添加内标的多重实时荧光pcr反应体系中,用以测定该pcr体系的扩增情况和内标的扩增情况,其扩增结果如图4所示。
结果表明,含猫流感病毒和猫疱疹病毒目标基因的质粒dna浓度在1.6×104拷贝/μl~1.6×107拷贝/μl时,内标的扩增均没有受到影响。虽然当dna的浓度高于1.6×106拷贝/μl时,内标的扩增受到了一点抑制,但此时含猫流感病毒和猫疱疹病毒目标基因的质粒dna很容易被检测出,因此,在这种情况下内标的扩增并不重要,因为只有在内标和检测目标dna的扩增均受到抑制的情况下,才能说明pcr抑制物的存在,检测结果为假阴性,因而并不影响内标作为假阴性结果指示物的功能。同时,在添加了内标时,猫流感病毒和猫疱疹病毒的扩增曲线呈典型的s型,扩增效率很高,没有受到抑制,说明添加内标对实时荧光pcr扩增体系中猫流感病毒和猫疱疹病毒的扩增没有任何影响。
上述实施例可广泛运用于荧光定量pcr内标的制备,所制备的内标序列与动物病原微生物基因组非同源,保证了内标扩增的特异性。本发明成功构建的内标组合物可用于单一或多种病原微生物的检测,有助于检测时显示假阴性的发生,在确保检测准确性与高效性的同时,大大降低检测成本,为满足临床检测提供有效可靠的技术手段。
以上对本发明的具体实施例进行了详细描述,但其只作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
序列表
<110>湖南冠牧生物科技有限公司
<120>一种内标组合物及其在检测动物病原微生物中的应用
<160>5
<170>siposequencelisting1.0
<210>1
<211>200
<212>dna
<213>内标基因(artificialsequence)
<400>1
ggcagcgtgcagctcgccgaccactaccagcagaacacccccatcggcgacggccccgtg60
ctgctgcccgacaaccactacctgagcacccagtccgccctgagcaaagaccccaacgag120
aagcgcgatcacatggtcctgctggagttcgtgaccgccgccgggatcactctcggcatg180
gacgagctgtacaagtaaag200
<210>2
<211>20
<212>dna
<213>引物ic-f(artificialsequence)
<400>2
gaccactaccagcagaacac20
<210>3
<211>19
<212>dna
<213>引物ic-r1(artificialsequence)
<400>3
gaactccagcaggaccatg19
<210>4
<211>20
<212>dna
<213>引物ic-r2(artificialsequence)
<400>4
cttgtacagctcgtccatgc20
<210>5
<211>22
<212>dna
<213>引物ic-p(artificialsequence)
<400>5
agcacccagtccgccctgagca22
- 还没有人留言评论。精彩留言会获得点赞!