一种3-芳基-2-丙炔-1-醇类衍生物及其制备方法与流程
本发明属于有机合成领域,具体涉及一种3-芳基-2-丙炔-1-醇类衍生物及其制备方法。
背景技术:
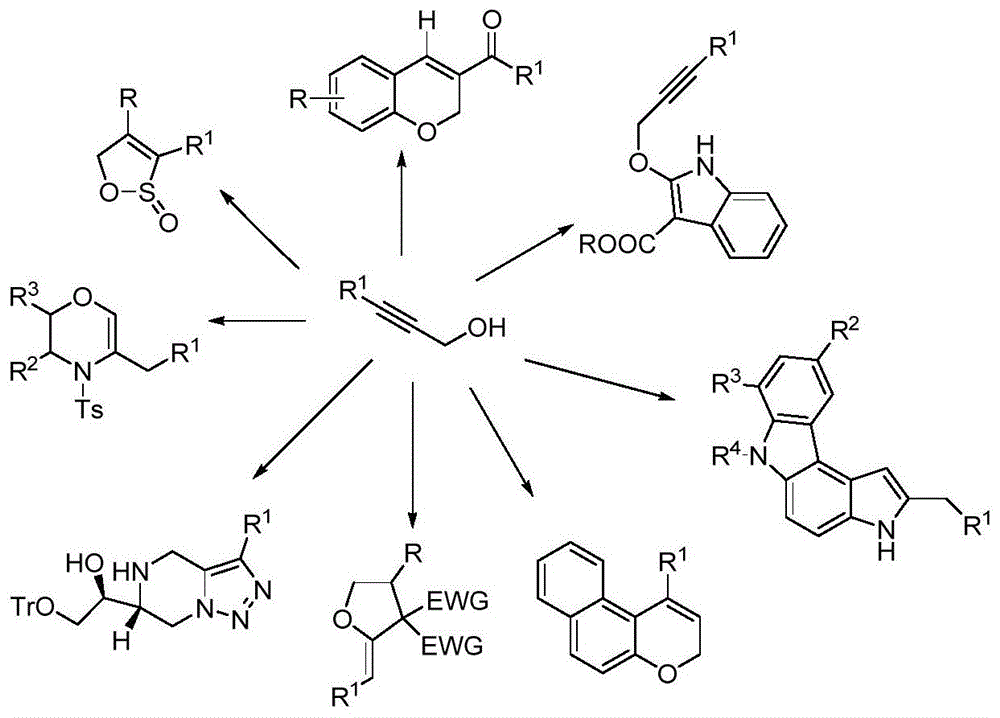
3-芳基-2-丙炔-1-醇类衍生物作为有机合成中的重要原料或中间体,用途十分广泛,在各种类型的有机合成中都是有用的构建基块,尤其是不同杂环骨架(见说明书附图图1)和复杂化合物(见说明书附图图2)的构建基块。如在农药和医药方面,其广泛用于合成各种生物活性和药用活性的大分子物质,见说明书附图图3,物质1是一种止痛药;物质2是天然分子白茅烯,具有生物活性;物质3是一种蛋白激酶抑制剂,拟用于治疗癌症,糖尿病和阿尔茨海默氏病。因此,3-芳基-2-丙炔-1-醇类衍生物的合成具有着重要的意义。
3-芳基-2-丙炔-1-醇类衍生物是生物活性化合物中重要的结构单元,因此,人们发展了许多制备它们的合成方法,主要有以下4类合成方法:(1)、传统的合成方法主要是使用强碱如烷基锂、二烷基锌、或有机镁试剂来激活末端炔烃,因此,这些方法的底物范围十分狭窄,而且需要低温零下78度条件;(2)、使用芳基卤化物和炔丙基醇衍生物合成芳基取代的炔丙基醇,但仅实验室条件下使用,因为商用终端炔丙基醇衍生物不是很多,导致底物的范围并不广泛,同时该反应需要使用贵金属pd做催化剂,成本昂贵;(3)、在较为温和的条件下,炔基硅烷和银化合物被用作炔基源与醛反应,但这些试剂必须制备,使一步反应变为了比较费力的两步反应,增加了反应步骤,这在一定程度上不符合绿色化学及原子经济的相关要求;(4)、铜催化多聚甲醛和末端炔反应的合成方案受到广泛关注,然而,它仍然需要苛刻的反应条件,如需要强碱和高温。综上所述,3-芳基-2-丙炔-1-醇类衍生物的现有合成方法大多在金属催化条件下进行,反应条件苛刻,需要强碱、高温(100℃)、或者低温零下78℃等条件。因此,寻求一种绿色高效、条件温和、方法简单、操作方便且产率高的方法是合成3-芳基-2-丙炔-1-醇类衍生物亟待解决的问题。
从原料的来源来看,传统上,各种甲醛的来源,如多聚甲醛、三恶烷、甲醛水溶液(福尔马林),或单体形式的气态甲醛。然而,它们的使用有几个缺点:多聚甲醛和三恶烷的反应性相对较低;多聚甲醛在有机溶剂中溶解度低,阻碍了工业规模反应,不适合大规模生产;福尔马林不适用于与容易质子化的亲核试剂反应;单体形式的气态甲醛为剧毒气体。因此,寻求一种简单、安全、用途广泛的甲醛源也是亟待解决的问题。
技术实现要素:
本发明的目的是要解决现有3-芳基-2-丙炔-1-醇类衍生物合成需要高温、金属催化、强碱或低温等条件导致的耗能高、成本高且污染环境的问题,而提供一种3-芳基-2-丙炔-1-醇类衍生物及其制备方法。
一种3-芳基-2-丙炔-1-醇类衍生物的结构式为:
一种3-芳基-2-丙炔-1-醇类衍生物的制备方法,是按以下步骤完成的:
一、将芳基乙炔类化合物、铵盐、碱、水和有机溶剂依次加入三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解1h~3h,得到反应混合物;
二、将反应混合物倒入盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
本发明的有益效果为:
一、本发明提供了一种3-芳基-2-丙炔-1-醇类衍生物,该衍生物具有潜在的生物活性和巨大的研究价值;本发明提供的衍生物具有较高的研究价值,可以用作药物先导化合物的筛选用以及供生物活性测试研究等科研单位使用;本发明亦可以作为合成其他含有3-芳基-2-丙炔-1-醇类衍生物的原料来使用;
二、本发明提供了一种简洁的一步法合成3-芳基-2-丙炔-1-醇类衍生物的方法,该方法解决了现有3-芳基-2-丙炔-1-醇类衍生物合成需要高温、金属催化、强碱或低温等条件导致的耗能高、成本高且污染环境的问题,寻求到了一条绿色高效、条件温和(室温)、方法简单、操作方便且产率高的合成3-芳基-2-丙炔-1-醇类衍生物的路线,为3-芳基-2-丙炔-1-醇类衍生物的工业化发展奠定了一定的基础;
三、本发明提供了一种极为简单、安全、用途广泛的方法,用于在有机溶剂中原位生成甲醛,该方法避免了气态甲醛的使用。本发明所使用的芳基三甲基铵三氟甲磺酸盐,可原位生成甲醛,为首次报道,具有创新性,是一种非常有用的和通用的3-芳基-2-丙炔-1-醇类衍生物合成方法。
附图说明
图1为3-芳基-2-丙炔-1-醇类衍生物可构建的不同杂环骨架的示意图;
图2为3-芳基-2-丙炔-1-醇类衍生物可构建的不同复杂化合物的示意图;
图3为利用3-芳基-2-丙炔-1-醇类衍生物合成的各种生物活性和药用活性的大分子物质,物质1是一种止痛药,物质2是天然分子白茅烯,物质3是一种蛋白激酶抑制剂;
图4为实施例1所得3-芳基-2-丙炔-1-醇类衍生物的1hnmr谱图。
图5为实施例1所得3-芳基-2-丙炔-1-醇类衍生物的13cnmr谱图。
具体实施方式
为了使本发明的技术方案及优点更加清楚明白,以下结合实施例对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
具体实施方式一:本实施方式是一种3-芳基-2-丙炔-1-醇类衍生物的结构式为:
具体实施方式二:本实施方式与具体实施方式一不同点是:所述的
具体实施方式三:本实施方式一种3-芳基-2-丙炔-1-醇类衍生物的制备方法,是按以下步骤完成的:
一、将芳基乙炔类化合物、铵盐、碱、水和有机溶剂依次加入三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解1h~3h,得到反应混合物;
二、将反应混合物倒入盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
具体实施方式四:本实施方式与具体实施方式一至三之一不同点是:步骤一中所述的芳基乙炔类化合物的结构式为:
具体实施方式五:本实施方式与具体实施方式一至四之一不同点是:所述的
具体实施方式六:本实施方式与具体实施方式一至五之一不同点是:步骤一中所述的电解的电流为恒电流,数值为10ma~20ma;步骤一中所述的芳基乙炔类化合物、铵盐、水和碱的摩尔比为1:1.8:7:2;步骤一中所述的芳基乙炔类化合物与有机溶剂的摩尔比为1mmol:20ml。其它步骤与具体实施方式一至五相同。
具体实施方式七:本实施方式与具体实施方式一至六之一不同点是:步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;步骤一中所述的碱为碳酸钠、氢氧化钠或乙醇钠;步骤一中所述的有机溶剂为无水n,n-二甲基甲酰胺或无水n,n-二甲基乙酰胺。其它步骤与具体实施方式一至六相同。
具体实施方式八:本实施方式与具体实施方式一至七之一不同点是:步骤一中所述的铵盐为n,n,n-三甲基苯铵三氟甲基磺酸盐或n,n,n-三甲基苄铵三氟甲基磺酸盐;所述的n,n,n-三甲基苯铵三氟甲磺酸盐的结构式为
本实施方式中n,n,n-三甲基苯铵三氟甲基磺酸盐的反应式为:
本实施方式中n,n,n-三甲基苄铵三氟甲基磺酸盐的反应式为:
具体实施方式九:本实施方式与具体实施方式一至八之一不同点是:步骤二中所述的盐水为饱和盐水;步骤二中所述的电源为恒电位仪,购买自上海昕瑞仪器仪表有限公司,恒电位仪型号:djs-292b(c)恒电位仪;步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa。其它步骤与具体实施方式一至八相同。
具体实施方式十:本实施方式与具体实施方式一至九之一不同点是:步骤二中所述的反应混合物与盐水的体积比为4:15;步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:(2~5)。其它步骤与具体实施方式一至九相同。
采用以下实施例验证本发明的有益效果:
实施例1:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol3-乙炔基苯甲醚、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例1的反应方程式为:
实施例1制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为72.0%,其1hnmr、13cnmr谱图分别为附件1中的图4、图5。
图4为实施例1所得3-芳基-2-丙炔-1-醇类衍生物的1hnmr谱图。
图5为实施例1所得3-芳基-2-丙炔-1-醇类衍生物的13cnmr谱图。
核磁数据分析为:1hnmr(400mhz,chloroform-d)δ7.21(t,j=8.0hz,1h),7.03(d,j=7.6hz,1h),6.97(s,1h),6.88(dd,j=8.3,2.5hz,1h),4.49(s,2h),3.79(s,3h),1.99(br,1h)。13cnmr(101mhz,chloroform-d)δ159.2,129.4,124.2,123.5,116.5,115.0,87.0,85.6,55.2,51.6。
实施例2:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol4-叔丁基苯基乙炔、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例2的反应方程式为:
实施例2制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为72.6%,核磁数据分析为:1hnmr(400mhz,chloroform-d)δ7.46–7.34(m,4h),4.53(s,2h),1.86(br,1h),1.35(s,9h)。13cnmr(101mhz,chloroform-d)δ151.7,131.4,125.3,119.4,86.5,85.8,51.7,34.7,31.1。
实施例3:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol1-乙炔基-2-氟苯、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例3的反应方程式为:
实施例3制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为94.9%,核磁数据分析为:1hnmr(400mhz,chloroform-d)δ7.43(td,j=7.4,1.8hz,1h),7.35–7.27(m,1h),7.13–7.02(m,2h),4.53(s,2h),1.96(br,1h).
13cnmr(101mhz,chloroform-d)δ162.8(d,j=251.5hz),133.6,130.2(d,j=8.0hz),123.9(d,j=3.7hz),115.5(d,j=20.9hz),111.1(d,j=15.6hz),92.4(d,j=3.3hz),79.0,51.6.
实施例4:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol1-氯-2-乙炔基苯、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例4的反应方程式为:
实施例4制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为95.4%,核磁数据分析为:1hnmr(400mhz,chloroform-d)δ7.47(dd,j=7.5,1.9hz,1h),7.39(dd,j=7.9,1.3hz,1h),7.23(dtd,j=19.9,7.5,1.6hz,2h),4.56(s,2h),2.21(br,1h)。13cnmr(101mhz,chloroform-d)δ135.8,133.5,129.5,129.2,126.4,122.4,92.4,82.3,51.6。
实施例5:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol1-乙炔基-4-甲基苯、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例5的反应方程式为:
实施例5制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为79.1%,核磁数据分析为:1hnmr(600mhz,chloroform-d)δ7.33(d,j=7.9hz,2h),7.11(d,j=7.8hz,2h),4.49(s,2h),2.34(s,3h),1.98(br,1h)。13cnmr(151mhz,chloroform-d)δ138.59,131.53,129.03,119.38,86.50,85.75,51.62,21.43。
实施例6:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol3-乙炔基吡啶、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例6的反应方程式为:
实施例6制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为83.6%,其核磁数据分析为:1hnmr(400mhz,chloroform-d)δ8.78(dd,j=2.2,0.9hz,1h),8.51(dd,j=5.0,1.7hz,1h),7.74(dt,j=7.9,1.9hz,1h),7.30–7.26(m,1h),4.72(br,1h),4.51(s,2h)。13cnmr(101mhz,chloroform-d)δ152.1,148.3,139.1,123.4,120.3,92.2,81.5,50.9。
实施例7:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol3-乙炔基噻吩、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例7的反应方程式为:
实施例7制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为74.3%,其核磁数据分析为:1hnmr(400mhz,chloroform-d)δ7.45(dd,j=3.0,1.1hz,1h),7.25(dd,j=5.0,3.0hz,1h),7.10(dd,j=5.0,1.1hz,1h),4.47(s,2h),2.06(br,1h)。13cnmr(101mhz,chloroform-d)δ129.9,129.2,125.5,121.6,86.9,80.9,51.6.。
实施例8:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol2-乙炔基萘、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例8的反应方程式为:
实施例8制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为53.2%,其核磁数据分析为:1hnmr(400mhz,chloroform-d)δ8.01–7.93(m,1h),7.83–7.75(m,3h),7.53–7.45(m,3h),4.56(s,2h),2.06(br,1h)。13cnmr(101mhz,chloroform-d)δ132.9,131.8,128.4,128.1,127.8,126.9,126.6,119.9,87.6,86.1,51.8。
实施例9:一种3-芳基-2-丙炔-1-醇类衍生物的制备方法是按以下步骤完成的:
一、将0.2mmol4-乙炔联苯、0.36mmoln,n,n-三甲基苯铵三氟甲基磺酸盐、0.4mmol碳酸钠、1.4mmol水、4mln,n-二甲基乙酰胺依次加入25ml三颈烧瓶中,再在三颈烧瓶中安装两个铂板电极,将两个铂板电极接通电源,在室温、空气气氛和搅拌条件下电解2h,得到反应混合物;
步骤一中所述的电解的电流为15ma恒电流;
步骤一中所述的两个铂板电极的尺寸均为10×10×0.1mm,两个铂板电极之间的距离为2cm;
二、将反应混合物倒入15ml盐水中,再使用乙酸乙酯萃取3次,合并有机层;再使用无水na2so4对合并的有机层进行干燥,最后进行减压蒸馏去除溶剂,得到粗产物;
步骤二中所述的盐水为饱和盐水;
步骤二中所述的电源为恒电位仪;
步骤二中所述的减压蒸馏的温度为35℃,压力为0.1mpa;
三、将粗产物通过硅胶柱色谱法纯化,其中,洗脱液为乙酸乙酯/石油醚的混合液,得到3-芳基-2-丙炔-1-醇类衍生物。
步骤三中所述的乙酸乙酯/石油醚的混合液中乙酸乙酯与石油醚的体积比为1:5。
实施例9的反应方程式为:
实施例9制备的3-芳基-2-丙炔-1-醇类衍生物的纯度为99%,产率为64.9%,其核磁数据分析为:1hnmr(400mhz,chloroform-d)δ7.61–7.57(m,2h),7.57–7.49(m,4h),7.48–7.42(m,2h),7.39–7.34(m,1h),4.53(s,2h),1.98(br,1h)。13cnmr(101mhz,chloroform-d)δ141.3,140.3,132.2,128.9,127.8,127.1,121.5,87.9,85.7,51.8。
以上所述的实施例只是本发明的一种较佳的方案,并非对本发明作任何形式上的限制,在不超出权利要求所记载的技术方案的前提下还有其它的变体及改型。
- 还没有人留言评论。精彩留言会获得点赞!