一种制备(S)-3-氨基-2-苄基丙酸甲酯的方法与流程
本发明涉及药物中间体合成技术领域,尤其涉及一种制备(s)-3-氨基-2-苄基丙酸甲酯的方法。
背景技术:
β-氨基酸作为一类重要的化合物,在药物合成以及蛋白质修饰等方面有着很多用途。合成这类分子的难点在于手性控制,(s)-3-氨基-2-苄基丙酸甲酯作为比较基础的手性β-氨基酸,目前在药物合成以及蛋白质修饰等方面有着很多用途。
现有的制备方法有:
1)手性拆分。包括结晶法、化学拆分法、萃取拆分法,这一方法的缺点是损失至少一半的原料,大部分拆分效率不高;
2)生物酶拆分法。这一方法稳定性较差,不利于应用于大规模生产(专利公开号wo2005085462a1、cn106191147a);
3)色谱拆分法。这一方法好处在于简单通用,但限制也很明显,设备成本高,制备量少,不适合大规模制备;
4)手性辅基诱导的不对称合成。这类虽然可以直接地构建手性中心,但是需要同等当量的手性辅基,也需要比较苛刻的条件,比如二(三甲基硅基)氨基锂(lihmds)、二异丙基氨基锂(lda)等金属试剂,不利于大量生产。
5)不对称氢化等金属催化的不对称合成。现阶段已工业化的不对称反应中氢化约占到70%(专利公开号cn102249833b),难点是寻找合适的手性催化剂,以及提高催化剂的催化效率。相较于传统的贵金属催化,有机小分子催化具有无重金属毒性以及反应条件温和等特点。
发明人已经发展了混合酸酐和保护苄基胺为起始原料,经过氮杂环卡宾(n-heterocycliccarbene,nhc)催化,得到各种β-氨基酸及其衍生物的方法(专利申请号cn201610715437.2)。
因此,本领域的技术人员致力于开发一种实用高效的手性制备(s)-3-氨基-2-苄基丙酸甲酯的方法。
技术实现要素:
有鉴于现有技术的上述缺陷,本发明所要解决的技术问题是如何实用且高效地手性制备(s)-3-氨基-2-苄基丙酸甲酯。
为实现上述目的,本发明提供了一种制备(s)-3-氨基-2-苄基丙酸甲酯的方法,所述方法包括以下步骤:
步骤1、将式1所示化合物与式2所示化合物经过氮杂环卡宾催化后利用甲醇进行淬灭反应,得到式3所示化合物;
步骤2、将所述式3所示化合物经兰尼镍催化进行氢化还原,得到式4所示化合物;
其中,步骤1中所述氮杂环卡宾的结构通式如下:
x为氢、溴和硝基中的一种,
r1为氢、甲基、乙基和异丙基中的一种,
r2为氢、甲基、乙基和异丙基中的一种。
进一步地,所述方法还包括:
步骤3、将所述式4所示化合物与氯化氢气体反应,得到所述式4所示化合物的盐酸盐。
进一步地,所述氮杂环卡宾为下列之一:
进一步地,在有机溶剂中,存在碱基,将所述式1所示化合物与所述式2所示化合物经过所述氮杂环卡宾催化。
进一步地,所述有机溶剂选自由二氯甲烷、二氯乙烷、乙酸乙酯、四氢呋喃、dmf和1,4-二氧六环组成的组。
进一步地,所述碱基选自由碳酸氢钠、碳酸钠、碳酸钾、三乙胺、dbu和dipea组成的组。
进一步地,在所述步骤1中,所述式1所示化合物、式2所示化合物和氮杂环卡宾的摩尔比为1:1-1.5:0.05-0.3。
进一步地,所述式1所示化合物、碱基和甲醇的摩尔比为1:0.2-1.5:1-3。
进一步地,在所述步骤2中,所述式3所示化合物和兰尼镍的摩尔比为1:0.1-1。
进一步地,在所述步骤3之前,利用乙酸乙酯对所述式4所示化合物进行萃取。
本发明的有益效果是原料简单易得、后处理简单,且对映体比率(enantiomericratio,e.r.)值和总收率良好,可以为工业化生产提供重要参考。
附图说明
图1是本发明的制备路线图。
具体实施方式
以下参考说明书附图介绍本发明的多个优选实施例,使其技术内容更加清楚和便于理解。本发明可以通过许多不同形式的实施例来得以体现,本发明的保护范围并非仅限于文中提到的实施例。
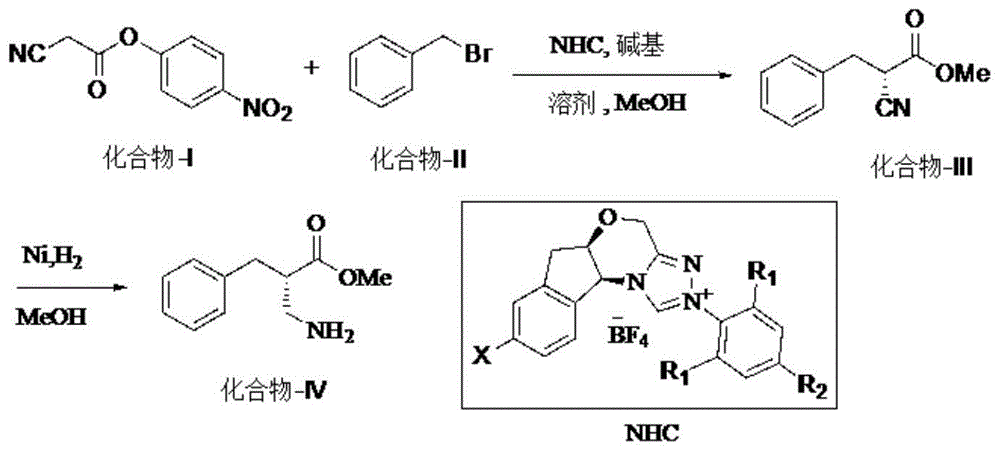
图1示出了本申请的(s)-3-氨基-2-苄基丙酸甲酯的制备路线。其中,该制备路线的每个结构式中的x和r代表以下基团:
x为氢、溴或者硝基;r1和r2为氢、甲基、乙基、异丙基。
本申请以氰乙酸对硝基苯酚酯以及苄基溴为起始原料,经过氮杂环卡宾催化得到手性氰基中间体,不分离直接兰尼镍催化氢化还原,得到(s)-3-氨基-2-苄基丙酸甲酯,经过简单的分离得到产品。本申请原料简单易得,后处理简单,e.r.值和总收率良好,可以为工业化生产提供重要参考。
具体包括以下步骤:
1、氰乙酸对硝基苯酚酯(化合物-ⅰ)的制备:
参考文献journalofheterocyclicchemistry,1981,18,1085–1087制备而得。
2、手性氰基(化合物-ⅲ)的制备:
向氰乙酸对硝基苯酚酯(化合物-i)溶剂中,加入氮杂环卡宾(nhc)催化剂前体、苄基溴(化合物-ⅱ)以及碱基(base),反应温度为0-40℃,监控化合物-ⅰ消失后,加入甲醇淬灭反应,得到化合物-ⅲ的反应液,为了避免得到的产物消旋,立即进行后一步反应。
3、化合物-ⅳ的制备:
上一步过柱得到的化合物-ⅲ的反应液,加入甲醇以及兰尼镍催化剂,氢气氛围下,加热回流至原料消失,过滤去除催化剂。冰水浴下,加入水稀释并用盐酸调节ph至3-4,随后减压去除甲醇,乙酸乙酯萃取2次水相。剩余水相用碳酸氢钠饱和液调节ph至中性后,乙酸乙酯萃取3次,合并后的有机相,干燥过滤后,通入干燥氯化氢气体,析出白色固体,过滤烘干后得到目标化合物-ⅳ的盐酸盐。
本申请的一些实施例的化合物-ⅲ的制备中,化合物-i、苄基溴的摩尔比为1:1-1.5,优选为1:1.1。化合物-i、氮杂环卡宾(nhc)催化剂前体的摩尔比为1:0.05-0.3,优选为1:0.1。化合物-i、碱基、甲醇的摩尔比为1:1-2:0.2-1.5:1-3,优选为1:1.1:1.2:2。优选反应温度为20℃,反应浓度为0.3m,反应时间为8-16小时。
所筛选的碱基为:碳酸氢钠、碳酸钠、碳酸钾、三乙胺、dbu以及dipea,优选的碱基为三乙胺。
所筛选的溶剂为:二氯甲烷、二氯乙烷、乙酸乙酯、四氢呋喃、dmf、1,4-二氧六环,优选的溶剂为四氢呋喃。
所筛选的nhc催化剂如下:
优选的nhc催化剂为nhc-ⅳ。
本申请的一些实施例的化合物-ⅳ的制备中,以化合物-ⅲ上一步收率100%计算,化合物-ⅲ、兰尼镍的摩尔比为1:0.1-1,优选为1:0.2,加氢反应温度为50-70℃,反应压力为1个大气压,优选甲醇浓度为0.3m,反应时间12-24小时。
实施例1
化合物-ⅴ的制备,由已知化合物-ⅰ和化合物-ⅱ制备而来,具体制备方法如下:
室温下,氮气保护下,向25ml圆底烧瓶中,加入氰乙酸对硝基苯酚酯(化合物-ⅰ)(0.60g,2.91mmol)、干燥的四氢呋喃(10ml)、苄基溴(化合物-ⅱ)(0.55g,3.2mmol)以及氮杂环卡宾催化剂前体(nhc-ⅳ)(140mg,0.29mmol)。冰水浴下,保持内温在5℃左右,慢慢加入三乙胺(tea)(0.32g,3.2mmol),并保持此温度搅拌8小时后,加入甲醇(0.24ml,5.82mmol),室温搅拌2h后。得到的化合物-ⅲ反应液直接后面一步反应。
上述得到的化合物-ⅲ反应液,加入甲醇(10ml),90%兰尼镍(38mg,0.58mmol),氢气球置换3次气体,加热回流,反应时间12小时。薄层层析(thinlayerchromatography,tlc)检测反应完成后,过滤去除催化剂,冰水浴下,加入10ml水稀释,随后盐酸调节ph至3-4,减压去除甲醇,乙酸乙酯萃取2次水相。剩余水相用碳酸氢钠饱和液调节ph至中性后,乙酸乙酯萃取3次,合并后的有机相,干燥过滤后,通入干燥氯化氢气体,析出白色固体,过滤烘干后得到目标化合物-ⅳ的盐酸盐0.45g,两步总收率为67%。1hnmr(400mhz,cd3od)7.42-7.14(m,5h),3.72(s,3h),3.25-2.83(m,5h).13cnmr(100mhz,cd3od)174.4,138.8,130.2,129.7,128.3,52.7,46.0,41.2,37.0.hrms(esi,m/z):[m-cl]+calcd.forc11h16no2194.11756,found:194.11745,hplcanalysis:97:3e.r.。
实施例2
将实施例1中的nhc催化剂变换为nhc-ⅶ,以及碱基变换为碳酸氢钠也能得到比较好的结果:
室温下,氮气保护下,向25ml圆底烧瓶中,加入氰乙酸对硝基苯酚酯(化合物-ⅰ)(0.60g,2.91mmol)、干燥的四氢呋喃(10ml)、苄基溴(化合物-ⅱ)(0.55g,3.2mmol)以及氮杂环卡宾催化剂前体(nhc-ⅶ)(135mg,0.29mmol)。冰水浴下,保持内温在5℃左右,慢慢加入碳酸氢钠(0.59g,3.2mmol),并保持此温度搅拌8小时后,加入甲醇(0.24ml,5.82mmol),室温搅拌2h后。得到的化合物-ⅲ反应液直接后面一步反应。后面操作与实施例1操作一致,得到目标化合物-ⅳ的盐酸盐0.41g,两步总收率为61%,核磁和质谱与实施例1一致,hplcanalysis:94:6e.r.。
以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!