一种天然PPARγ受体激动剂及其制备方法和应用与流程
一种天然ppar
γ
受体激动剂及其制备方法和应用
技术领域
[0001]
本发明属于核受体超家族的配体激活转录因子制备技术领域,涉及一种天然pparγ受体激动剂,还涉及上述激活受体激动剂的制备方法和应用。
背景技术:
[0002]
过氧化物媒体增殖物激活受体(pparγ),pparγ高表达于脂肪细胞、脾、肾上腺和结肠中,低表达于肝脏、肾、脑、骨骼肌等组织。其中pparγ研究最为深入,是目前抗糖尿病药物筛选最常用的靶标之一。其在多种细胞的增殖与分化过程中起着重要的调节作用,激活pparγ具有促进脂肪细胞分化,抗动脉粥样硬化、减少炎症的发生、治疗癌症等作用。目前,寻求无副作用或副作用小的新型pparγ激动剂及其作用机制研究正成为抗糖尿病药物研究的热点之一。寻求天然的pparγ激动剂,可以有效地克服目前临床上广泛使用的抗糖尿病药物的副作用,具有较大的科学意义、应用价值和社会效应。目前,发现的天然pparγ受体激动剂不是很多,主要有amorfrutins、多炔、甾醇类等化合物具有较好的pparγ激动活性,且副作用小。amorfrutins类化合物是从我国传统中药甘草中发现的具有较强胰岛素增敏效果的pparγ激动剂,无体重增加等副作用,是一种具有开发前景的天然抗糖尿病药物;从羌活的地下部分得到的聚乙炔类化合物falcarindiol,对pparγ受体具有潜在激动活性,可以诱导前脂肪细胞3t3-l1的分化和提高人单核细胞(thp-1)的胆固醇代谢水平。这些研究结果进一步表明,从天然药物中更有可能发现具有副作用小的新型pparγ受体激动剂类先导化合物但是,目前从我国传统中药材中寻求pparγ受体激动剂的研究仍然较少。
技术实现要素:
[0003]
本发明的第一个目的是提供一种天然pparγ受体激动剂,该化合物在10μm的浓度下,其萤火虫荧光素酶活性和内对照海肾荧光素酶活性的相对平均强度与pparγ受体的全激动剂罗格列酮的相当,其可以为研发新型天然的抗糖尿病药物提供一种先导化合物。
[0004]
本发明的第二个目的是提供上述受体激动剂的制备方法。
[0005]
本发明的第三个目的是提供上述受体激动剂在抗糖尿病药物中的应用。
[0006]
本发明所采用的第一种技术方案是,一种天然pparγ受体激动剂,受体激动剂为从自山茱萸中提取的熊果酸。
[0007]
本发明所采用的第二种技术方案是,一种天然pparγ受体激动剂的制备方法,具体按照以下步骤实施:
[0008]
步骤1:制备山茱萸提取物;
[0009]
步骤2:将步骤1得到的山茱萸提取物进行分离纯化得到受体激动剂。
[0010]
本发明所采用的第二种技术方案的特点还在于,
[0011]
步骤1的制备过程如下:
[0012]
步骤1.1:将山茱萸阴干粉碎之后,以75%的工业乙醇提取得总浸膏;
[0013]
步骤1.2:将步骤2.1得到的浸膏充分分散到水里,用3倍体积的石油醚脱脂,得石
油醚部位;
[0014]
步骤1.3:将步骤1.2得到的萃余相加入3倍体积的乙酸乙酯萃取,旋干得到乙酸乙酯部位。
[0015]
步骤2具体为:
[0016]
步骤2.1:将步骤1.3得到的乙酸乙酯提取物溶解,加入200-300目的硅胶搅拌均匀混合,乙酸乙酯提取物和硅胶的质量比为1:1,待溶剂挥发完样品干燥后,用研钵把样品磨成粉末备用;
[0017]
步骤2.2:在色谱柱依次加入不同浓度梯度的石油醚-乙酸乙酯及乙酸乙酯-甲醇进行梯度洗脱,每个浓度冲6l溶剂;
[0018]
步骤2.3:将步骤2.2中各个浓度梯度的流出液用旋转蒸发仪转干;
[0019]
步骤2.4:对步骤2.3中各个部分采用tlc薄层板点板,紫外灯,硫酸乙醇显色情况,对分离得到的组分合并,最终合并为6个组分,标记为fr.1:pe:ea=80:20和60:40,27.7g、fr.2:pe:ea=40:60,2.2g、fr.3:pe:ea=20:80,135.7g、fr.4:ea 100%,299.4g、fr.5:ea:meoh=80:20,156.6g、fr.6:ea:meoh=60:4,0
→
0:100,15.1g;
[0020]
步骤2.5:将fr.1和fr.2合并后,以硅胶为固定相,采用梯度洗脱剂进行洗脱并收集,收集后使用旋转蒸发仪转干,分别收集并作好标记,将各个组分进行tlc点板,并在紫外灯下观察点板结果,再使用硫酸乙醇或碘试剂显色,根据tlc薄层色谱板显示的比移值,将各所含化合物相近的组分进行合并,得到16个子组分:fr.1-1、fr.1-2、fr.1-3、fr.1-4、fr.1-5、fr.1-6、fr.1-7、fr.1-8、fr.1-9、fr.1-10、fr.1-11、fr.1-12、fr.1-13、fr.1-14、fr.1-15、fr.1-16;
[0021]
步骤2.6:对fr.1-6先应用反相高效液相色谱进行梯度洗脱分离,以含0.1%的甲酸的水和甲醇为洗脱剂,按此时间程序85:15
→
65:35 in 40min,v/v,接着65:35
→
0:100 in 20min 4ml/min设置的比例进行梯度洗脱,在保留时间为23.4min时收集到一个组分,将此组分进一步应用反相氨基柱色谱进行梯度洗脱分离:以含0.1%的甲酸的水和甲醇为洗脱剂,按此时间程序80:20
→
65:35 in 40min,v/v,接着65:35
→
0:100 in 20min 4ml/min得到化合物1,所述化合物1经核磁谱图和质谱鉴定为熊果酸。
[0022]
步骤2.2中石油醚与乙酸乙酯的体积比依次为80:20、60:40、40:60、20:80和100%的乙酸乙酯,所述乙酸乙酯和甲醇的体积比依次为80:20、60:40、40:60、20:80和100%的甲醇。
[0023]
步骤2.3中洗脱剂为pe-etoac和etoac-meoh不同浓度的混合溶液,按每个梯度增加20%的极性较大的溶剂的比例增大混合溶液的极性。
[0024]
本发明所采用的第三种技术方案是:一种天然pparγ受体激动剂在抗糖尿病药物中的应用。
[0025]
本发明所采用的第三种技术方案的特点还在于,
[0026]
受体激动剂在10μm的浓度下,其萤火虫荧光素酶活性和内对照海肾荧光素酶活性的相对平均强度与pparγ受体的全激动剂罗格列酮的相当。
[0027]
本发明的有益效果是:从我国传统中草药山茱萸中发现了一种对pparγ受体γ具有显著激动活性的化合物,为研发新型天然抗糖尿病药物提供了新的先导化合物和科学依据。
附图说明
[0028]
图1是本发明一种天然pparγ受体激动剂熊果酸的核磁共振氢谱;
[0029]
图2是本发明一种天然pparγ受体激动剂熊果酸的核磁共振碳谱;
[0030]
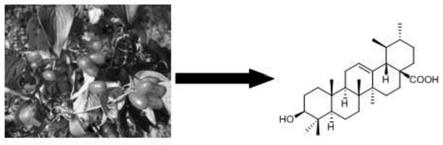
图3是本发明一种天然pparγ受体激动剂熊果酸的化学结构式。
具体实施方式
[0031]
下面结合附图和具体实施方式对本发明进行详细说明。
[0032]
本发明一种天然pparγ激动剂的制备方法,具体按照以下步骤实施:
[0033]
步骤1:植物提取
[0034]
步骤1.1:将山茱萸阴干粉碎之后,以75%的工业乙醇提取得总浸膏;
[0035]
步骤1.2:将浸膏充分分散到水里,用3倍体积的石油醚脱脂,得石油醚部位;
[0036]
步骤1.3:将步骤1.2的萃余相加入3倍体积的乙酸乙酯萃取,旋干得到乙酸乙酯部位。
[0037]
步骤2:分离纯化
[0038]
步骤2.1:将步骤1.3得到的乙酸乙酯提取物加入乙酸乙酯、石油醚、甲醇等溶剂使其溶解,与干硅胶(200-300目)按质量比1:1的比例混合,搅拌均匀,以使样品能够完全被硅胶吸附,待溶剂挥发完样品干燥后,用研钵把样品磨成粉末备用;
[0039]
步骤2.2:选择适宜大小的玻璃色谱柱(10
×
56cm),先将干硅胶装入玻璃色谱柱中,之后用石油醚不断地冲洗色谱柱以便排尽气泡,通过干法上样,将硅胶吸附好的样品载入硅胶色谱柱中,以pe-ea,ea-meoh不同极性的混合溶液作为洗脱剂,对硅胶柱色谱进行梯度洗脱,每个比例冲6l溶剂;
[0040]
步骤2.3:将各个比例的流出液用旋转蒸发仪转干,得10个组分;
[0041]
步骤2.4:对各个部分根据tlc薄层板点板,紫外灯,硫酸乙醇显色情况,对分离得到的组分合并,最终合并为6个组分,标记为fr.1(pe:ea=60:40,27.7g)、fr.2(pe:ea=40:60,2.2g)、fr.3(pe:ea=20:80,135.7g)、fr.4(ea 100%,299.4g)、fr.5(ea:meoh=80:20,156.6g)、fr.6(ea:meoh=60:4,0
→
0:100,15.1g)。
[0042]
步骤2.5:将fr.1和fr.2合并(即pe:ea=40:60、60:40,共29.9g),用甲醇、乙酸乙酯等各种有机溶剂溶解后,与硅胶按2:1的比例混合进行拌样,采用干法装柱,以硅胶为固定相,再用石油醚不停地洗脱排尽气泡后,采用干法上样,以pe-etoac(100:0
→
0:100)和etoac-meoh(100:0
→
0:100)不同浓度的混合溶液作为洗脱剂,按每个梯度增加20%的极性较大的溶剂(etoac或甲醇)的比例增大混合溶液的极性,每个比例配制4l溶液,对色谱柱进行梯度洗脱。对不同极性的洗脱剂冲洗下来的溶液收集,使用旋转蒸发仪转干,分别收集于样品瓶(25ml)中,并作好标记。将各个组分进行tlc点板,并在紫外灯下观察点板结果,再使用硫酸乙醇或碘试剂显色,根据tlc薄层色谱板显示的比移值,将各所含化合物相近的组分进行合并,得到16个子组分(记为fr.1-1至fr.1-16)。fr.1-1(0.25g)、fr.1-2(0.15g)、fr.1-3(2.1g)、fr.1-4(0.06g)、fr.1-5(0.12g)、fr.1-6(0.18g)、fr.1-7(0.68g)、fr.1-8(2.83g)、fr.1-9(0.22g)、fr.1-10(0.54g)、fr.1-11(2.32g)、fr.1-12(1.67g)、fr.1-13(1.28g)、fr.1-14(0.44g)、fr.1-15(0.2g)、fr.1-16(0.06g)。
[0043]
步骤2.6:对fr.1-6(0.18g)先应用反相高效液相色谱(rp-c18 column;5μm;10
×
250mm)进行梯度洗脱分离(以含0.1%的甲酸的水和甲醇为洗脱剂,按此时间程序85:15
→
65:35 in 40min,v/v,接着65:35
→
0:100 in 20min 4ml/min)设置的比例进行梯度洗脱,在保留时间为23.4min时收集到一个组分,该组分总量有30mg。将此组分进一步应用反相氨基柱色谱(ymc-pack nh2,s-5,10
×
250mm)进行梯度洗脱分离(以含0.1%的甲酸的水和甲醇为洗脱剂,按此时间程序80:20
→
65:35in 40min,v/v,接着65:35
→
0:100 in 20min4ml/min)的化合物1(15mg,t
r
=18.09min)。
[0044]
步骤3:化合物结构鉴定
[0045]
步骤3.1:白色结晶粉末,且tlc薄层板点板显示只有1个点;
[0046]
步骤3.2:根据1h、
13
c nmr(c5d5n,400mhz)核磁谱图数据(见图1和图2),结合质谱[m-h]-m/z=455.2,并与文献对比,确定化合物1为熊果酸,其化学式如图3所示。
[0047]
步骤4:pparγ受体激动剂活性测试
[0048]
步骤4.1:将化合物1配制成不同浓度的溶液(1.0,3.0和10.0μm)存储于冰箱中备用;
[0049]
步骤4.2:将ht29(人结肠癌细胞)细胞接种在dmem(含10%胎牛血清)培养基上,在co2培养箱中培养,培养条件为37℃,5%co2,相对湿度90%;
[0050]
步骤4.3:将步骤4.2的ht29细胞融合形成致密的单层细胞后,用0.125%胰蛋白酶作用3~5min,此操作仍在37℃条件下,若发现细胞间隙增大了,则吸出消化液终止消化;
[0051]
步骤4.4:用dmem培养基清洗1次,去除死亡细胞的碎片以及残余胰蛋白酶;加入dmem完全培养基5ml,反复吹打,制成细胞悬液,接种于25ml培养瓶中,按1:2的比例分瓶培养,采用倒置显微镜每日观察细胞生长情况。
[0052]
步骤4.5:将0.2μg报告基因质粒ppre-tk-luc:addgene或对照质粒phrl-cmv加入到25μl的i reduced serum medium中,混合均匀;
[0053]
步骤4.6:将0.5μl lipofectamin2000稀释到25μl的i reduced serum medium中,轻轻混匀,静置5min;
[0054]
步骤4.7:将质粒溶液加入到lipofectamin2000溶液中,轻轻混匀,静置20min;
[0055]
步骤4.8:将细胞培养板中的培养液吸出,每孔重新加入50μl新鲜的培养基(含10%胎牛血清的dmem);将质粒/lipo混合溶液加入到细胞培养孔中,每孔50μl;
[0056]
步骤4.9:在转染6h后换液,换成不含质粒/lipo混合溶液,得到稳定表达细胞,继续扩增培养;
[0057]
步骤4.10:培养好的细胞按照8000细胞/孔接种到96孔细胞培养板中,37℃、5%co2培养12h后按溶剂对照组(dmso)、阳性对照组(ros)、不同浓度的待测样品进行处理后,置于co2培养箱内孵育24小时后,弃上清,用ph7.3的pbs充分洗涤细胞后,每孔加入100l细胞裂解液裂解细胞,收集细胞裂解产物进行荧光素酶报告基因检测;
[0058]
步骤4.11:在96孔板上进行的细胞实验中,无论是阴性对照、阳性对照或待测药物,每个样品在同一浓度下,都需要设置3个复孔,并重复进行3次独立的实验,只有当每次实验获得的结果相近时,才证明所获实验数据真实有效;
[0059]
步骤4.12:取20μl细胞裂解产物,采用双荧光素酶报告基因检测试剂盒进行目的报告基因萤火虫荧光素酶及内对照中海肾荧光素酶活性检测,pparγ的转录活性以萤火虫
荧光素酶活性和内对照海肾荧光素酶活性的比值表示,如下表1所示,为熊果酸对pparγ受体的激动活性检测结果,由表1可知:熊果酸在10μm时的相对荧光强度与pparγ的全激动剂罗格列酮的相当,表明该化合物具有较强的pparγ激动活性。通过上述方式,本发明一种对pparγ受体具有显著激动活性的三萜类化合物,可为研发新型天然的抗糖尿病药物提供了潜在的先导化合物。
[0060]
表1.熊果酸对pparγ受体的激动活性
[0061][0062][0063]
目前发现的pparγ激动剂除了合成的激动剂外,还有内源性激动剂。主要包括:(1)前列腺素衍生物,如15d-pgj2,pgj,pgd2等;(2)脂肪酸及其衍生物,如亚油酸、二十碳五烯酸等。合成的pparγ全激动剂,如以罗格列酮(rosiglitazone,rog)为代表的胰岛素增敏剂四氢噻唑烷二酮类化合物(thiazolidinediones,tzds)是也是迄今唯一被应用于临床的pparγ受体激动剂(曲格列酮、罗格列酮、吡格列酮等),能显著提高糖尿病患者(尤其是ii型糖尿病)的胰岛素敏感性、降低血糖和血脂、改善心血管和高血压等疾病的症状。但能引起患者水肿、体重增加和心衰等严重的副作用。相对于合成的外源性激动剂,内源性配体虽安全性好,但活性较弱。因此,从天然药物中发现安全有效的pparγ配体已受到研究者广泛关注。本发明一种天然pparγ受体激动剂的制备方法,其特征在于:从我国常用中草药“山茱萸”的果实中发现了一种对pparγ受体具有显著激动活性的天然激动剂,该研究结果不仅可为开发新型的抗糖尿病药物提供先导化合物,也为我国民间药物山茱萸的深入开发、利用提供可靠的科学依据。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1