一种胺基中间体的精制方法与流程
[0001]
本发明属于药物合成技术领域,具体涉及一种胺基中间体的精制方法。
背景技术:
[0002]
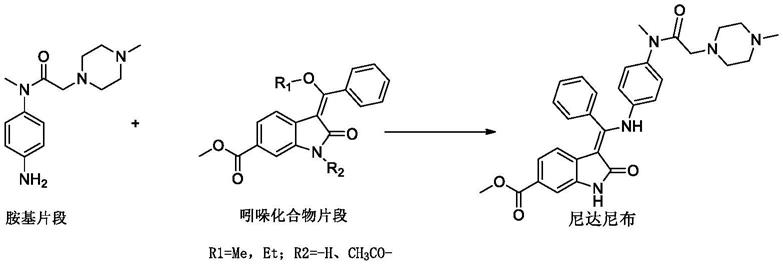
乙磺酸尼达尼布,全名:1h-吲哚-6-羧酸,2,3-二氢-3-[[[4-[甲基[(4-甲基-1-哌嗪基)乙酰基]氨基]苯基]氨基]苯基亚甲基]-2-氧-,甲酯,(3z)-,乙磺酸盐(1:1),英文名:nintedanib esilate,是由勃林格殷格翰公司开发的一种治疗特发性肺纤维化药物。商品名:ofev,于2014年10获得fda批准上市。在尼达尼布的合成方法中,胺基片段和吲哚化合物片段是其两个重要的中间体。
[0003][0004]
现有技术(cn101883755a)、j.med.chem.2009,52,4466
–
4480及合成化学2015年第23卷第8期,763~766报道的胺基片段(式(i))的合成方法如下:
[0005][0006]
研究发现,合成式(i)所示化合物时,现有技术一般采用催化氢化,(pd/c)、水合肼-三氯化铁或铁粉还原等,但采用上述方法来制备式(i)所示化合物时会有少量式(ii)所示化合物(即杂质1)和还原中间态(即杂质2)残留。
[0007]
根据appendix 1of eur 23844en,jrc scientific and technical reports,2009中列举的警示结构(structural alert),查询后,发现上述两个杂质均具有警示结构,且与原料药结构无关,无致突变数据。根据ich-m7的分类依据,将其归为第3类(即潜在遗传毒性杂质)。
[0008]
ich m7指导原则6危害评估要素评估指出,对于具有足够的致癌性数据的杂质(1类),应按该物质的特异性数据来推导可接受限度。对具有警示结构,与原料药结构无关,无致突变性数据的杂质(3类)和致癌性未知的已知致突变物(2类)均要控制在合适的ttc限度以下。
[0009]
在ich m7指导原则7.1基于ttc的可接受摄入量指出,一个致突变杂质每天每人摄
入1.5μg时其风险被认为是可以忽略的(终生暴露情况下理论患癌风险小于十万分之一),这一值可以通用于大部分药物,作为可接受控制限度的默认值。该方法一般用于长期治疗用(>10年)药物中存在的且无致癌数据(2类和3类)的致突变杂质。
[0010]
根据乙磺酸尼达尼布软胶囊说明书信息可知,该药品的最大日剂量为300mg(以碱计),换算成盐后其最大日剂量为360mg。
[0011]
根据ich-m7“遗传毒性杂质限度指南”中提到由ttc推算的原料药中遗传毒性杂质的浓度限度(ppm)计算公式为:
[0012][0013][0014]
即杂质1和杂质2作为潜在遗传毒性杂质,在乙磺酸尼达尼布原料药中需要控制其在4ppm以下。但现有技术均未报道对杂质1和杂质2在胺基中间体和成品中作为潜在遗传毒性杂质的控制策略,这在药品质量控制和药品使用上存在较高风险。
[0015]
为减轻或降低潜在遗传毒性杂质残留风险和成品因多次精制而造成的物料损耗,需要开发一种合适的胺基中间体的精制方法,将上述杂质控制在合理范围内,以确保成品中这两杂质的限度在4ppm以下。这将在药品使用及保障人民用药安全方面具有重大的意义和价值。
技术实现要素:
[0016]
本发明的目的在于解决上述技术问题,提供一种制备高质量胺基中间体的方法,可以将胺基中间体中的杂质1和2含量降低至4ppm以下,从而保证乙磺酸尼达尼布中的杂质1和2含量在4ppm以下,使其符合药用标准。
[0017]
本发明的技术方案是通过如下方式实现的:
[0018]
式(i)所示的胺基中间体的精制方法,
[0019][0020]
包括:式(i)所示的化合物与溶剂混合,并进行析晶步骤,其中,溶剂选自四氢呋喃和甲苯的混合溶剂、四氢呋喃和甲基叔丁基醚的混合溶剂。
[0021]
更进一步地,所述溶剂选自四氢呋喃和甲苯的混合溶剂,其中,四氢呋喃和甲苯的体积比为1:1~8:1,优选3:1~6:1,最优选5:1。
[0022]
更进一步地,所述溶剂选自四氢呋喃和甲基叔丁基醚的混合溶剂,其中,四氢呋喃和甲基叔丁基醚的混合溶剂中,四氢呋喃和甲基叔丁基醚的体积比为1:1~8:1,优选2:1~5:1,最优选3:1。
[0023]
进一步地,所述式(i)化合物和溶剂的质量体积比(g/ml)为1:20~1:40,优选1:25~1:30。
[0024]
进一步地,所述精制方法还包括式(i)化合物与溶剂混合后,加热溶解。
[0025]
进一步地,所述精制方法中的析晶步骤为降温析晶。
[0026]
更进一步地,所述降温析晶的温度选自10℃~50℃,优选15℃~40℃,最优选20℃~30℃。
[0027]
进一步地,所述降温析晶步骤包括二步降温析晶。
[0028]
更进一步地,所述第2步降温析晶的温度选自-10℃~30℃,优选-5℃~20℃,最优选0℃~15℃。
[0029]
进一步地,所述降温析晶的降温时间控制在2-15小时,优选4~12小时,最优选5~8小时。
[0030]
进一步地,第2步降温析晶是通过搅拌析晶,析晶时间1~15小时,优选1~10小时,最优选2~5小时。
[0031]
本申请中发明人惊奇的发现,通过本申请技术方案精制得到的胺基中间体,能够得到高收率、高纯度的胺基中间体的同时,获得基因毒杂质1和基因毒杂质2均显著下降的目标产物,可以将胺基中间体中的杂质1和2含量降低至4ppm以下,使得乙磺酸尼达尼布符合药用标准。
具体实施方式
[0032]
为了更好地理解本发明的内容,下面结合具体实施例作进一步说明。但是,这些实施例不能构成对本发明的保护范围的限制。
[0033]
胺基中间体和乙磺酸尼达尼布的液质检测方法见下表1:
[0034]
表1
[0035]
[0036][0037]
实施例1:式(i)化合物的制备
[0038]
将n-(4-硝基苯氨基)-n-甲基-2-氯-乙酰胺(式(ⅲ)化合物)80g加入到360ml乙酸乙酯中,加热至40℃,滴加n-甲基哌嗪86.9g。50℃搅拌反应2小时,降温至室温,加入50ml水洗涤。用异丙醇500ml稀释有机层,加入钯炭(10%)5g。室温氢化,过滤,60℃减压浓缩除去溶剂,得到浓缩物78g。
[0039]
实施例2:式(i)化合物的精制考察
[0040]
将式(i)化合物11g加入到乙酸乙酯-甲苯(1:7)330ml,升温,搅拌溶解后,6小时内逐步降温至20~30℃,继续降温至10℃,搅拌析晶4小时,过滤,80℃真空干燥,质量收率65.5%。
[0041]
实施例3:式(i)化合物的精制考察
[0042]
将式(i)化合物11g加入到乙酸乙酯330ml,升温,搅拌溶解后,6小时内逐步降温至20~30℃,继续降温至10℃,搅拌析晶4小时,过滤,80℃真空干燥,质量收率62.2%。
[0043]
实施例4:式(i)化合物的精制考察
[0044]
将式(i)化合物浓缩物11g加入到乙腈-甲基叔丁基醚(1:1)11ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率73.5%,纯度97.6%。
[0045]
实施例5:式(i)化合物的精制考察
[0046]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲苯(12:1)300ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率75.5%,纯度72.2%。
[0047]
实施例6:式(i)化合物的精制考察
[0048]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲基叔丁基醚(12:1)300ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率71.0%,纯度70.0%。
[0049]
实施例7:式(i)化合物的精制考察
[0050]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲苯(5:1)11ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率78.0%,纯度75.2%。
[0051]
实施例8:式(i)化合物的精制考察
[0052]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲苯(5:1)300ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率93.5%,纯度99.8%。
[0053]
实施例9:式(i)化合物的精制考察
[0054]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲苯(1:1)220ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率92.2%,纯度99.5%。
[0055]
实施例10:式(i)化合物的精制考察
[0056]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲苯(8:1)440ml中,升温,搅拌溶解后,5小时内逐步降温至20~30℃,继续降温至5℃,搅拌析晶2小时,过滤,80℃真空干燥,质量收率90.8%,纯度99.6%。
[0057]
实施例11:式(i)化合物的精制考察
[0058]
将式(i)化合物浓缩物11g加入到四氢呋喃-甲基叔丁基醚(3:1)280ml,升温,搅拌溶解后,8小时内逐步降温至20~30℃,继续降温至0℃,搅拌析晶5小时,过滤,80℃真空干燥,质量收率92.5%,纯度99.4%。
[0059]
实施例12:尼达尼布的制备
[0060]
将实施例7制备的式(i)化合物4.3g与3-[甲氧基(苯基)亚甲基]-2-氧化二氢吲哚-6-甲酸甲酯5g于甲醇36ml和n,n-二甲基甲酰胺9ml的混合物,加热至回流。回流7小时后,将反应液冷却至0℃,保温搅拌2小时。过滤,干燥,得到7.1g尼达尼布,纯度99.7%。
[0061]
实施例13:乙磺酸尼达尼布的制备
[0062]
将实施例7制备的尼达尼布6g加入到甲醇40ml和0.5ml水中,加热至60℃。将乙磺酸水溶液1.75g加入反应液中,加入40ml异丙醇过程中,控温在55~60℃。加毕,降温至0~5℃,保温搅拌2小时,过滤,干燥,得到7.0g乙磺酸尼达尼布,纯度99.5%。
[0063]
试验例1:杂质检测
[0064]
对实施例1的式(i)化合物浓缩物,实施例2-11精制的胺基中间体,以及由实施例2-11精制的胺基中间体制备得到的乙磺酸尼达尼布进行有关物质检测,主要检测杂质1和杂质2的含量,结果见下表2:
[0065]
表2潜在遗传毒性杂质精制效果对比
[0066][0067][0068]
由上表可见,现有技术报道的精制方法虽然可以降低潜在基因毒性杂质1和2的含量,但是与ich-m7“遗传毒性杂质限度指南”中提到由ttc推算的原料药中遗传毒性杂质的浓度限度(4.17ppm)相比相差很大,溶剂的种类、溶剂比例,以及式(i)化合物与溶剂用量的不同显著影响胺基中间体的纯化效果,本发明的精制方法可以将胺基中间体中杂质1和杂质2的含量控制在1.5ppm以下,且乙磺酸尼达尼布中杂质1和杂质2的含量在0.3ppm以下。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1